
Vol. 35, n.º 1, 2002
| REVISTA
ESPAÑOLA DE Vol. 35, n.º 1, 2002 |
Armando Martínez Martínez1, Alberto Rábano Gutiérrez2
1 Departamento de Anatomía Patológica, Sección de Neuropatología, Hospital Clínico de San Carlos, Madrid. 2 Laboratorio de Neuropatología, Fundación Hospital Alcorcón, Madrid.
INTRODUCCIÓN
Los efectos de la ingesta excesiva de alcohol sobre el sistema nervioso (SN) son múltiples pués, además del efecto tóxico directo que el etanol tiene sobre el SN, en el alcoholismo crónico se asocian con gran frecuencia otros procesos que, en definitiva, son los causantes de los trastornos neurológicos más comunes asociados a esta adicción, así como una mayor incidencia de diversas enfermedades neurológicas. Así, en el alcoholismo encontramos:
Defiencias nutricionales.
Afectación de órganos cuya patología repercute secundariamente sobre el SN (cirrosis hepática fundamentalmente).
Posible existencia de tóxicos contaminentes en las bebidas alcohólicas.
Alta incidencia de traumatismos craneales, a veces no bien documentados, que provocan hematomas subdurales; de hecho, el 50% de ellos está asociado a alcoholismo (1).
Mayor frecuencia de hemorragias subaracnoideas e intraparenquimatosas, en general de tipo lobar, cuyo riesgo aumenta en relación directa con la cantidad de etanol ingerida. Si bien el consumo leve de alcohol parece disminuir la incidencia de accidente vascular cerebral, el consumo severo aumenta hasta 2,5 veces el riesgo de padecerlo (2).
Mayor incidencia de procesos infecciosos.
Situación etiológica tan compleja hace que en el alcoholismo sea difícil deslindar las lesiones atribuibles directamente al efecto del etanol de la patología asociada y secundaria (3,4). A todo esto debemos sumar el hecho de que en mismo enfermo coinciden con cierta frecuencia diversos tipos de patología asociada con el alcoholismo. Por otro lado, está perfectamente probado el efecto teratogénico del alcohol, responsable del síndrome alcohólico fetal.
A continuación se describe la anatomía patológica de los procesos más frecuentemente asociados con la ingesta excesiva de etanol, con independencia de su etiología.
INTOXICACIÓN ALCOHÓLICA AGUDA
La intoxicación alcohólica aguda produce grado variable de estimulación del SN (regocijo, excitación, desinhibición, locuacidad, agresividad, irritabilidad, descoordinación), pero si es intensa, puede seguirse de una fase depresiva (somnolencia, estupor) que puede conducir a coma y, en casos severos, a muerte por depresión cardio-respiratoria. En estos casos la autopsia muestra congestión, edema y hemorragias petequiales difusas. De forma ocasional pueden encontrarse hemorragia o infarto masivos, usualmente en el contexto de hipertensión arterial o arteriosclerosis preexistentes.
ENCEFALOPATÍA DE WERNICKE (EW) Y SÍNDROME DE WERNICKE-KORSAKOFF (SK)
La EW está producida por la deficiencia de tiamina (vitamina B1), vitamina hidrosoluble y termolábil que no se sintetiza en hígado y que se almacena sólo en pequeñas cantidades en el organismo, por lo que es importante la ingesta continuada de alimentos que la contengan. Desempeña un papel importante como coenzima en el metabolismo de los carbohidratos y en el ciclo del ác. cítrico, por lo que la deficiencia de vitamina B1 afecta al metabolismo oxidativo.
Wernicke describió este síndrome en tres enfermos, dos alcohólicos y una joven con estenosis pilórica por ingestión de ác. sulfúrico. Estos casos reflejan los grupos de riesgo más importantes en Occidente: alcoholismo crónico y malnutrición severa secundaria a trastornos gastrointestinales. También se ha descrito en alimentación intravenosa prolongada inadecuada, grapado gástrico por obesidad mórbida, hemodiálisis crónica, SIDA, etc.
La incidencia de lesiones características de EW en el alcoholismo varía entre el 1 y el 3% en las grandes series de autopsias (5,6,7,8), y sólo el 10% de los casos había sido diagnosticado en vida, lo que explica que la prevalencia clínica de este proceso sea mucho menor. La EW debe considerarse como una urgencia clínica, ya que el retraso en el tratamiento con tiamina en las fases iniciales puede hacer que este proceso, esencialmente reversible, se transforme en un cuadro patológico irreversible. El término psicosis de Korsakoff (SK) se refiere a un síndrome amnésico, que suele cursar junto con la EW; el tratamiento adecuado produce recuperación total en el 25% de los casos y parcial sólo en el 50%.
Las alteraciones neuropatológicas de la EW varían según el estadio y la severidad del proceso. Los cuerpos mamilares están afectados prácticamente siempre (fig. 1), pero en los casos más severos hay tambien lesiones en estructuras cercanas a las paredes del tercer ventrículo (hipotálamo, región periventricular del tálamo), sustancia gris periacueductal, suelo del cuarto ventrículo y con menor frecuencia otras áreas (5,6,7,8). Se admiten tres estadios, agudo, subagudo y crónico, en el desarrollo de la enfermedad, pero es frecuente que se repitan los episodios y que se solapen lesiones en diferentes fases evolutivas.

Fig. 1. Encefalopatía de
Wernicke. Imagen macroscópica correspondiente a un corte coronal a nivel de
cuerpos mamilares. Estos muestran coloración herrumbrosa (oscura) por depósito
de pigmento hemosiderínico (Cortesía del Dr. J. Escalona Zapata, Servicio de
Anatomía Patológica, Hospital Gregorio Marañón).
Macroscópicamente, en los casos agudos pueden verse hemorragias petequiales en las zonas señaladas, pero con frecuencia las lesiones no son detectables a menos que se realice estudio microscópico (7); sólo en raras ocasiones las hemorragias son grandes y llamativas. En enfermos que han sobrevivido a un episodio agudo o han sufrido varios ataques, los cuerpos mamilares muestran grado variable de atrofia y al corte muestran color parduzco (fig. 1), así como las lesiones periventriculares y periacueductales (si existen), con una estrecha banda de tejido nervioso preservado inmediatamente adyacente a la cavidad ventricular. Se han descrito lesiones unilaterales, pero pueden tener origen isquémico, por afectación del territorio de la arteria coroidea posterior, o estar producidas por degeneración transneuronal tras lesión hipocámpica ipsilateral (9). La atrofia de los cuerpos mamilares, característica de la encefalopatía de Wernicke crónica, puede demostrarse con técnicas de neuroimagen (10).
Los hallazgos microscópicos varían tambien según el estadio y severidad de la enfermedad y son similares en todas las regiones afectadas. En el 25% de los casos sólo se encuentran alteraciones tras la realización de estudios histológicos por lo que, aún en ausencia de datos clínicos característicos, es importante la selección de muestras, al menos de los cuerpos mamilares, en grupos de riesgo (11).
En la fase aguda se observan edema, hemorragias petequiales y desmielinización. Las neuronas no parecen ser el blanco inicial de las lesiones y, salvo en raras ocasiones muestran algunos cambios (12), lo que explica la remisión de los signos y síntomas clínicos si se instaura precozmente tratamiento con tiamina. Recientemente se ha descrito vacuolización y degeneración neuronal en el tálamo en la fase aguda de la EW (13).
Si no se administra vitamina B1, continúa la evolución del proceso y en poco tiempo se establece la fase subaguda; en 1-2 días se observa hipertrofia endotelial y yemas capilares que en la segunda semana alcanzan el grado máximo de proliferación (fig. 2 y 3); a veces se observa necrosis, a menudo bilateral y simétrica, más frecuente en el tálamo. Al cabo de unos días aparece tambien reacción astrocítica. A menudo hay destrucción mielínica, con aparición de macrófagos espumosos, y axonal, responsable quizá de la presencia de cromatolisis neuronal en los cuerpos mamilares.

Fig. 2. Encefalopatía de Wernicke.
Microfotografía panorámica de un cuerpo mamilar en el que se observa aumento
considerable de su vascularización. Hematoxilina-Eosina.

Fig. 3. Encefalopatía de Wernicke.
Detalle de la figura anterior que muestra, junto a la gran proliferación
vascular, intensa hiperplasia endotelial en los vasos neoformados.
Hematoxilina-eosina.
En la fase crónica los casos leves pueden mostrar en las regiones afectadas poco más que gliosis discreta y algunos macrófagos con hemosiderina; sin embargo, si los cambios han sido más severos existe pérdida neuronal, gliosis severa que imparte aspecto esponjoso a la zona central de los cuerpos mamilares y abundantes macrófagos con hemosiderina, testigos de las hemorragias previas. Los vasos adquieren morfología habitual.
Las lesiones del tronco cerebral en la EW son del mismo tipo que las descritas en los cuerpos mamilares; los núcleos más afectados (8) son los vestibulares y oculomotores, entre otros muchos. En general las lesiones en el tronco cerebral son mucho más frecuentes en los casos agudos que en los crónicos (8), lo que concuerda con la remisión usualmente rápida de la oftalmoplejía y el nistagmus en las fases agudas tras tratamiento con tiamina.
Aparte de las lesiones intersticiales y vasculares de la EW, se han realizado numerosos estudios en otras regiones encefálicas para tratar de aclarar las bases neuropatológicas del SK. Los hallazgos han sido muchas veces discordantes debido a la valoración fundamentalmente cualitativa de las lesiones. Inicialmente se atribuyó este proceso a la lesión de los cuerpos mamilares, pero pronto se centró la atención en la afectación de núcleos talámicos específicos, debido a la existencia de casos de psicosis de Korsakoff asociados a hemorragias y neoplasias en esta región. El tálamo se afecta en el 53-100% de los casos de SK, sobre todo el núcleo medial dorsal, por lo que esta lesión fué la que primero se relacionó con la amnesia (6). Sin embargo, algunos enfermos con este síndrome muestran lesiones en cuerpos mamilares y estructuras de la línea media del tálamo, manteniendo intacto el núcleo medio dorsal (8,14,15). En un estudio morfológico cuantitativo realizado en casos de EW con SK en alcohólicos (16), se encuentra que tanto el núcleo anterior del tálamo como el dorsomedial muestran lesiones antes de que los enfermos con EW desarrollen SK. Las lesiones talámicas difieren de las típicas intersticiales y vasculares de la EW por la existencia en tálamo de lesiones y pérdida de neuronas, en contraste con la relativa conservación neuronal en el resto de las áreas clásicamente afectadas (13,17).
Estudios cuantitativos y morfométricos en el núcleo basal de Meynert aportan datos discordantes. En algunos trabajos no se encuentra pérdida de neuronas, ni en alcohólicos sin demencia (18), ni en el SK (15). Sin embargo, en un estudio morfométrico detallado realizado en 7 alcohólicos, 2 de ellos con SK (16), había pérdida significativa de neuronas en la región CH4 del núcleo de Meynert, tanto en enfermos con EW (22%) como en la SK (25%); aunque no existía relación causal entre la pérdida neuronal y la amnesia, sí parecía haberla con la pérdida de atención de algunos enfermos.
Las alteraciones en las olivas inferiores son frecuentes y comparten con las lesiones talámicas la pérdida neuronal y la conservación relativa del neuropilo y del endotelio capilar (8). Además, los núcleos olivares a menudo muestran degeneración transináptica secundaria a la degeneración del vermis cerebeloso, que tan frecuentemente se encuentra en el alcoholismo.
Se ha resaltado la importancia de la lesión del locus cerúleo y su vía noradrenérgica en el desarrollo de SK en el alcoholismo crónico. Inicialmente se describió pérdida significativa de neuronas en enfermos con amnesia severa (15), hallazgo que concuerda con la reducción significativa de noradrenalina y sus metabolitos en líquido cefalorraquídeo en el SK y con la mejora de la amnesia tras la instauración de tratamiento sustitutivo con noradrenalina. Sin embargo, en estudios cuantitativos posteriores en alcohólicos con EW, algunos con psicosis de Korsakoff y amnesia severa (19), no se han encontrado alteraciones significativas en este núcleo con respecto al grupo control.
Tampoco hay unanimidad de criterios en cuanto a la afectación de los núcleos del rafe. En un estudio cuantitativo realizado en el núcleo dorsal del rafe, cuya localización hace probable su daño en las lesiones periventriculares en el SK, no se encontraron alteraciones (15). Sin embargo, existen niveles bajos de metabolitos serotonérgicos en líquido cefalorraquídeo en alcohólicos con SK y el tratamiento para aumentar la actividad serotonérgica mejora la función mnésica. Además, en un estudio cuantitativo de los núcleos dorsal y medio del rafe con técnicas inmunohistoquímicas para neuronas serotonérgicas en 9 alholólicos con SK (4 de ellos con amnesia severa) y en 5 controles (20), se encontró pérdida significativa de neuronas serotonérgicas y de sus fibras en ambos núcleos en todos los pacientes alcohólicos.
De todo lo expuesto se desprende que es fundamental la utilización de técnicas cuantitativas y morfométricas adecuadas, así como uniformidad en la selección de muestras y una estrecha correlación anatomoclínica, para la valoración objetiva de los hallazgos encontrados.
PELAGRA
La pelagra o «mal de la rosa» se debe a la deficiencia de ácido nicotínico (vitamina B3, vitamina PP, niacina), necesario para la síntesis de NADH y NADPH. Normalmente se obtiene de la dieta (sobre todo de carne, hígado, salvado de arroz y levadura) pero tambien se sintetiza a partir del triptófano. En Occidente existe déficit dietético de ácido nicotínico en alcohólicos crónicos con problemas nutritivos graves (21,22) y en enfermos tratados con isoniacida (23), pero tambien aparece en vegetarianos estrictos, en la malabsorción intestinal y en la enfermedad de Hartnup (defecto hereditario de la absorción de triptófano, aminoácido precursor del ácido nicotínico). El término pelagra se refiere a la lesión cutánea, el más frecuente de los síntomas que constituían la triada típica que caracterizaba a la deficiencia endémica de vitamina B3: dermatitis, diarrea y demencia. La forma esporádica occidental carece a menudo de dermatitis y diarrea y puede ser difícil su diagnóstico; de hecho, en los casos asociados a alcoholismo la mayoría de las veces se diagnostica en la autopsia (21), pudiendo coincidir con otros procesos normalmente encontrados en esta adicción, como EW, enfermedad de Marchiafava-Bignami o ambos (22,24).
Desde el punto de vista neuropatológico (21,22,23,24,25), el cerebro es macroscópicamente normal y el principal y casi único hallazgo microscópico es la presencia de cromatolisis central en las neuronas de gran tamaño de todo el sistema nervioso central, sin lesiones asociadas en los axones, la glía ni la mielina. Las neuronas aparecen hinchadas, carentes de grumos de Nissl y con el núcleo lateralizado, aspecto de la reacción axonal clásica; sin embargo, los axones no muestran cambios, por lo que se cree que se trata de una lesión primaria. Las neuronas más afectadas son las pirámides de Betz del cortex motor prerrolándico, neuronas de varios núcleos del tronco cerebral (sobre todo en la protuberancia), motoneuronas anteriores de la médula espinal, células de Purkinje y células piramidales del hipocampo. La severidad de los cambios medulares varía de caso a caso y según el nivel estudiado (22), lo que podría explicar algunas discrepancias en los hallazgos.
ATROFIA CEREBRAL
En el curso del alcoholismo crónico aparece a veces deterioro intelectual global que se ha relacionado con la existencia de atrofia cerebral y agrandamiento ventricular, detectados en estudios de neuroimagen y necrópsicos, aunque se discute la relación de estos hallazgos con el efecto tóxico directo del alcohol (26,27). En el alcoholismo parece haber una disminución significativa del peso y volumen cerebral, sobre todo si se asocia SK (28,29). Análisis morfométricos han demostrado que la atrofia se debe sobre todo a reducción de volumen de la sustancia blanca hemisférica cerebral (30,31), aunque con resonancia magnética tambien se ha encontrado reducción del volumen cortical (32).
Estudios microscópicos cuantitativos realizados en alcohólicos crónicos (33) demuestran pérdida significativa de neuronas en el córtex frontal superior (área 8 de Brodmann), independientemente de la presencia o no de síndrome de Korsakoff o de cirrosis hepática. Sin embargo, otros trabajos morfométricos no encuentran diferencias en el número de neuronas entre el grupo de alcohólicos sin síndrome de Korsakoff y el grupo de control (34). En un estudio reciente se estudian volúmenes y contajes neuronales en ciertas regiones cerebrales en alcohólicos con EW, con síndrome de Korsakoff y no complicados con estos cuadros, relacionándolos con la antigüedad e intensidad del consumo de alcohol y comparándolos con un amplio grupo de control (27). En este trabajo se confirma la disminución de volumen de la sustancia blanca, en especial en el lóbulo frontal, en todos los alcohólicos, sobre todo en los casos con EW (con o sin psicosis de Korsakoff), y se demuestra pérdida de neuronas en la corteza de asociación frontal superior; además, tras la aplicación de estudios inmunohistoquímicos parece que la toxicidad afecta a neuronas piramidales no gabaérgicas. Es necesaria la realización de estudios con técnicas morfométricas y cuantitativas precisas de las mismas regiones cerebrales para poder contrastar los resultados de forma homogénea. El amplio espectro de cambios microscópicos en las neuronas corticales descrito clásicamente en el alcoholismo (hinchazón, atrofia pigmentaria, picnosis, pérdida de pirámides pequeñas) se considera actualmente inespecífico o de carácter artefactual.
ESCLEROSIS CORTICAL LAMINAR DE MOREL
Originalmente descrita por Morel en 1939 en alcohólicos crónicos con cuadro clínico de seudoparálisis cerebral y sintomatología similar al síndrome de Korsakof; microscópicamente mostraban pérdida neuronal y gliosis limitadas a la capa III de la corteza cerebral, sin alteraciones en ganglios basales ni cerebelo (35). Esto da lugar a la formación de una banda de aspecto esponjoso en la capa III del cortex cerebral, a veces tambien en la capa IV, que afecta especialmente a las regiones frontal lateral y temporal.
La esclerosis laminar de Morel suele ocurrir asociada a la enfermedad de Marchiafava-Bignami (36) y en general se considera que es secundaria a ésta. Sin embargo, se han descrito casos de esclerosis laminar cortical aislada, como manifestación neuropatológica única de alcoholismo crónico (37). Tambien hay casos asociados conjuntamente a enfermedad de Marchiafava-Bignami y mielinolisis pontina central (38) y a mielinolisis pontina y extrapontina no relacionados con alcoholismo (39).
DEGENERACIÓN CEREBELOSA ALCOHÓLICA
En alcohólicos crónicos se observa con frecuencia atrofia de la porción anterior del vermis superior y áreas adyacentes de los hemisferios cerebelosos (40). Su incidencia es muy elevada —casi el 27% de los alcohólicos y del 32 al 38,6% de los que presentan síndrome de Wernicke-Korsakoff (29,7)— y puede aparecer de forma aislada o asociada a otros procesos relacionados con el alcoholismo. Se afectan más los hombres que las mujeres. El cuadro clínico se instaura lentamente, con inestabilidad troncal, ampliación de la base de sustentación y marcha atáxica.
Macroscópicamente, existe adelgazamiento de las laminillas cerebelosas y ensanchamiento de los surcos en el vermis rostral (fig. 4) y, en menor grado, en la cara superior de los hemisferios cerebelosos en zonas adyacentes al vermis. Microscópicamente, las lesiones son más intensas en las crestas de las laminillas que en la profundidad de los surcos, al contrario de lo que ocurre en las lesiones hipóxicas. En las zonas afectadas existe pérdida de células de Purkinje (fig. 5) y pérdida parcheada variable de células granulares, atrofia de la capa molecular y gliosis de Bergmann (41,42); la pérdida de células de Purkinje es más marcada en alcohólicos con síndrome de Wernicke-Korsakoff (43). Con el método de Golgi se observa reducción de la arborización dendrítica y pérdida de espinas en las células de Purkinje restantes (44).
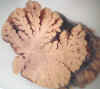
Fig. 4. Atrofia cerebelosa en el
alcoholismo crónico. Fotografía macroscópica de un corte sagital de vermis
cerebeloso, que muestra marcada atrofia de las laminillas en su parte superior,
con ensanchamiento de los surcos.

Fig. 5. Corte histológico de
vermis atrófico en el que existe pérdida masiva de células de Purkinje.
Hematoxilina-eosina.
La atrofia cerebelosa que ocurre en el alcoholismo crónico posiblemente guarda relación con el déficit de tiamina más que con el efecto tóxico directo del alcohol.
ENFERMEDAD DE MARCHIAFAVA-BIGNAMI (EMB)
Se trata de un proceso raro caracterizado por desmielinización central del cuerpo calloso. Fué descrito originalmente en alcohólicos italianos adictos a vino tinto «peleón» y malnutridos (45). Posteriormente se ha encontrado en mujeres, individuos de otras nacionalidades, en alcohólicos sin preferencias por un tipo determinado de bebidas (46) e incluso en no bebedores malnutridos (47). Puede haber diferencias regionales, mostrando al parecer mayor incidencia en Francia que en otros paises (23). Además de mostrar con frecuencia lesiones de esclerosis cortical laminar de Morel (36), la EMB se puede asociar a EW (46), a cambios morfológicos de tipo pelagroso en sistema nervioso central (23,25) e incluso a mielinolisis pontina central (48,39). Antes se diagnosticaba sólo en autopsia, pero actualmente puede detectarse con tomografía computarizada y resonancia magnética (49), que ha aportado datos importantes sobre la evolución de este proceso (50) y la existencia de casos de evolución benigna (51,52).
Desde el punto de vista neuropatológico (36,53) se caracteriza macroscópicamente por la presencia de una zona de desmielinización, descolorida e incluso parcialmente quística o colapsada, en la zona central del cuerpo calloso, con afectación generalmente máxima en la rodilla y el cuerpo (fig. 6). De forma característica la lesión está muy bien delimitada de la sustancia blanca adyacente conservada, manteniéndose dos capas periféricas de fibras mielínicas a lo largo de las caras dorsal y ventral de esta comisura. La zona de desmielinización puede estar circunscrita en una zona más o menos amplia del cuerpo calloso, pero puede extenderse ampliamente a la sustancia blanca hemisférica en forma de alas de mariposa (fig. 7). Tambien se han encontrado lesiones desmielinizadas, en general bilaterales y simétricas, en quiasma, comisura anterior y pedúnculos cerebelosos medios, así como en la sustancia blanca hemisférica. Microscópicamente existe pérdida extensa de la mielina con preservación relativa de los axones, disminución del número de oligodendrocitos y abundantes macrófagos cargados de lípidos en las fases iniciales. Los astrocitos muestran en general escasos cambios reactivos, pero en las lesiones más destructivas puede haber gliosis y formación de quistes. Los vasos en y alrededor de la lesión suelen mostrar proliferación e hialinización parietal (fig. 8). En un estudio de correlación radiológico-neuropatológica se encontraron depósitos masivos de hemosiderina en macrófagos y astrocitos en la pared de una lesión quística del cuerpo calloso, que se relacionó con un aumento transitorio del contraste tomográfico en la fase subaguda, probablemente por hemorragia (54).

Fig. 6. Enfermedad de
Marchiafava-Bignami. Corte coronal a nivel de la rodilla del cuerpo calloso en
el que se observa macroscópicamente una estría desmielinizada en la porción
central de la comisura, característica de este proceso.

Fig. 7. Enfermedad de
Marchiafava-Bignami. Corte histológico coronal de ambos hemisferios cerebrales,
teñido con técnica mielínica de azul luxol y violeta de cresilo (técnica de
Klüver-Barrera). Existe desmielinización de la porción central del cuerpo
calloso, netamente delimitada que, en este caso, se acompaña de pérdida
submasiva de la mielina en la sustancia blanca hemisférica.

Fig. 8. Enfermedad de
Marchiafava-Bignami. Detalle microscópico de la zona afectada que muestra
aspecto laxo por la pérdida mielínica y esclerosis de la pared vascular.
Tricrómico de Masson.
No se conoce la etiopatogenia de la EMB, pero suele asociarse con alcoholismo y malnutrición, aunque no todos los casos tienen historia de abuso de alcohol. Lesiones similares se pueden producir en la intoxicación crónica con cianuros. En estudios seriados en una mujer se vió resolución casi completa de las imágenes patológicas descubiertas con tomografía computarizada y de resonancia magnética tras tratamiento con complejo vitamínico B (50). Estudios radiológicos cuantitativos cuidadosos en alcohólicos crónicos sin EMB evidencian atrofia con adelgazamiento del cuerpo calloso, sobre todo en la rodilla y el cuerpo (55).
MIELINOLISIS PONTINA CENTRAL
Fué descrita en 1959 en 4 enfermos con malnutrición severa, tres de ellos alcohólicos crónicos (56). En casos publicados posteriormente se ha expandido el espectro de procesos subyacentes, habiéndose descrito en trastornos electrolíticos severos, enfermedades debilitantes crónicas, cirrosis hepática y trasplante hepático, uremia, leucemia, enfermedad pulmonar crónica, quemaduras graves, etc. Aunque es más frecuente en individuos de mediana edad, se ha descrito tambien en niños (57,58). A veces aparece junto con otros procesos asociados a alcoholismo crónico, sobre todo con encefalopatía de Wernicke, pero tambien con cambios pelagrosos e incluso con enfermedad de Marchiafava-Bignami (48,38). El diagnóstico clínico es difícil pero su sospecha puede corroborarse con técnicas de radiodiagnóstico (59,60), aunque las lesiones pueden pasar inadvertidas inicialmente (61). La tomografía computarizada y la resonancia magnética han permitido identificar las fases iniciales del proceso y casos con recuperación completa.
Neuropatológicamente, la lesión típica consiste en una zona triangular o romboidal descolorida en la porción central de la base de la protuberancia, desmielinizada y parcialmente necrótica (fig. 9), que puede incluso estar cavitada. Su tamaño varía de lesiones mínimas, detectables sólo en cortes histológicos (62), a lesiones extensas que afectan casi toda la superficie de corte transversal de la base del puente. Generalmente, queda intacto al menos un fino ribete mielínico en los bordes laterales y anterior de la protuberancia. Ocasionalmente las lesiones son bífidas, en alas de mariposa, por afectación de las porciones laterales, preservando la línea media del puente. La desmielinización suele ser máxima en las porciones medial y rostral de la protuberancia, con extensión variable al tegmento y, raras veces, en sentido rostral hacia el mesencéfalo. Caudalmente la desmielinización suele detenerse en la unión bulboprotuberancial. Sólo raras veces se afecta el bulbo raquídeo.
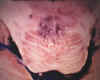
Fig. 9. Mielinolisis pontina
central. Fotografía macroscópica en la que se aprecia una zona parcialmente
necrosada en la porción central de la protuberancia (Cortesía del Dr. J. L.
Sarasa Corral, Servicio de Anatomía Patológica de la Fundación Jiménez
Díaz, Madrid).
Microscópicamente (fig. 10), la lesión corresponde a una zona bien delimitada de desmielinización con preservación de los axones y de los cuerpos neuronales dentro del área afectada, lo que distingue este proceso de un infarto. En las fases iniciales hay numerosos macrófagos espumosos pero escaso o nulo infiltrado inflamatorio perivascular. Los oligodendrocitos están muy reducidos en número o incluso faltan. Ocasionalmente, en el centro de las lesiones más severas se produce necrosis y cavitación, pero incluso entonces, en la zona periférica se reconocen neuronas conservadas. Alrededor de las lesiones se pueden encontrar axones fragmentados y formación de hinchazones argirófilas procedentes de los mismos. Las fibras más afectadas son las pontocerebelosas transversas, seguidas de las vías largas rostrocaudales. Inmunohistoquímicamente se ha descrito en las fases iniciales una disminución de tinción para proteina gliofibrilar ácida y en las lesiones antiguas se refiere una «distrofia astroglial» (63), sugierendo que estos cambios pueden ser importantes en la patogénesis de la MPC. En la zona lesionada y a su alrededor hay astrocitosis reactiva y ocasionales astrocitos tipo II de Alzheimer. El estudio ultraestructural está limitado por artefactos; se ha descrito dehiscencia intramielínica, con vacuolización y ruptura de las vainas (64,65), mecanismo diferente al de otras enfermedades desmielinizantes, como esclerosis múltiple, pero similar al que se ve en algunos procesos tóxicos y metabólicos (65).

Fig. 10. Mielinolisis pontina
central. Corte histológico de la protuberancia (caso anterior) teñido con la
técnica milínica de eriocromocianina y rojo nuclear. Se aprecia
desmielinización extensa bien delimitada en la región central del puente.
En el 10% de los casos, sobre todo en los más severos, se encuentran focos de desmielinización extrapontinos a nivel del estriado, cápsulas interna y externa, tálamo, cuerpos geniculados laterales, comisura anterior, fórnix y sustancia blanca de las laminillas cerebelosas (57,58,59,66,67,39).
La patogenia de la mielinolisis pontina central no se conoce completamente. En base a datos clínicos y experimentales, parece que guarda relación con trastornos del equilibrio hidroelectrolítico y en particular con hiponatremia, sobre todo si es crónica y cuando se corrige de forma excesivamente rápida o en exceso (68,69,70,71). Otra teoría apunta a la hiperosmolaridad o hipertonicidad relativa o absoluta, más que a la alteración específica de los niveles de sodio (67,72,73). Pero hay casos recientes que cuestionan este mecanismo y se han propuesto otras hipótesis, como hipofosfatemia (74). No se conoce la base de la afectación preferentemente pontina de la desmielinización. Algunos lo atribuyen a edema en áreas donde se interpone sustancia gris entre bandas de fibras mielínicas longitudinales y transversales (68), en las que una mayor proporción de células oligodendrogliales se encuentra en estrecha proximidad a la sustancia gris más vascularizada (73). El edema ocurriría por aumento de la permeabilidad de la barrera hemato-encefálica por lesión osmótica en el endotelio (72,75).
NEUROPATÍA ALCOHÓLICA
Es una de las formas más frecuentes de polineuropatía. Estudios electromiográficos revelan anomalías en el 93% de los alcohólicos, aunque muchos de ellos estén asintomáticos, a pesar de mostrar signos clínicos de neuropatía. Usualmente ocurre en el contexto de alcoholismo crónico, en general severo; se desarrolla insidiosamente y cursa como una neuropatía sensitivo-motora distal y simétrica. A veces hay disfonía por afectación vagal, así como síntomas autonómicos, aunque éstos suelen ser raros, aparte de la anhidrosis distal en los pies.
Desde el punto de vista neuropatológico la primera lesión que se evidenció fué desmielinización segmentaria (76). Luego se encontró degeneración axonal (77), demostrándose que la desmielinización segmentaria era consecuencia de un proceso degenerativo axonal distal (78,79,80). La severidad de las lesiones (depleción de fibras mielínicas, degeneración activa) guarda relación directa con la cronicidad del proceso.
El parecido anatomoclínico de la neuropatía alcohólica con la que ocurre en el beriberi y la frecuente asociación de deficiencias nutritivas en el alcoholismo crónico ha hecho pensar que la neuropatía alcohólica es el resultado del déficit de tiamina y no del efecto tóxico directo del alcohol, aunque éste no está excluido (79), pero no hay una demostración clara ni un modelo experimental animal satisfactorio.
Existen pocos datos con respecto a la neuropatía autónoma alcohólica. Muestreos con tests para la detección de alteraciones funcionales vegetativos (simpáticas y parasimpáticas) evidencian una proporción variable de alteraciones (81,82), sobre todo en los grupos de más edad, en mujeres y cuando existe enfermedad hepática establecida (81), así como un aumento de la mortalidad en los pacientes con neuropatía vagal (82). Los estudios morfológicos en el vago en alcohólicos crónicos son escasos. Un estudio morfométrico (73) demuestra en todos los casos estudiados una disminuciíon significativa de la densidad de fibras mielínicas en la parte distal del nervio y degeneración axonal compatible con neuropatía retrógrada, previamente descrita (69).
OTRAS LESIONES
Mielopatía alcohólica: La mayoría de los casos de mielopatía descrita en alcohólicos se ha relacionado con cirrosis severa y shunts portocava y de hecho han sido denominadas «mielopatías por shunt». Sin embargo, se han publicado 5 casos de mielopatía progresiva en pacientes alcohólicos bien nutridos y en los que la abstinencia detuvo la progresión del proceso, aunque sin mejoría (83). No existen estudios neuropatológicos sobre este cuadro.
Una alteración frecuente en la médula espinal de los alcohólicos es la degeneración de los cordones posteriores, secundaria a neuropatía periférica.
Miopatía alcohólica: En el curso del alcoholismo crónico se puede desarrollar una miopatía aguda (84) caracterizada morfológicamente por mionecrosis, presencia de vacuolas lipídicas en las fibras tipo 1, infiltrado inflamatorio y fenómenos regenerativos. Con el paso del tiempo se observa fibrosis, desaparecen las vacuolas lipídicas y hay atrofia de fibras tipo 2B (85).
También puede asociarse miositis con cuerpos de inclusión al alcoholismo (84).
Al parecer relacionada con la hepatopatía crónica, en el curso del alcoholismo existe aumento de fenómenos de distrofia neuroaxonal (86,87,88), así como presencia de glía tipo II de Alzheimer tipo II, como consecuencia del desarrollo de encefalopatía hepática.
Aunque se ha descrito la aparición de neuropatía óptica en el alcoholismo, posiblemente sea debida a déficit de vitaminas B o a contaminación de las bebidas alcohólicas con metanol.
SÍNDROME ALCOHÓLICO FETAL (SAF)
El SAF comprende un amplio espectro de lesiones, neurológicas y somáticas, debidas al efecto del alcoholismo materno sobre el feto. Se ha estimado su incidencia en 0,33/1.000 nacidos vivos; sin embargo, como sus efectos son difíciles de reconocer en el neonato y la disfunción del SN central puede no ser evidente hasta varios años después, la incidencia verdadera del SAF puede ser mucho más alta. En la actualidad se considera que es la causa más frecuente de retardo mental y de otros defectos del nacimiento, por encima del síndrome de Down o la espina bífida. Además de la cantidad de alcohol ingerida existen otros factores que intervienen en la vulnerabilidad al desarrollo de SAF, como diferencias genéticas del metabolismo del alcohol en la madre, asociación de otras drogas y patrón de la ingesta. En este sentido, las borracheras parecen ser más nocivas que la misma o mayor cantidad de alcohol consumida de forma repartida. Ni siquiera se sabe con certeza si la causa de las múltiples manifestaciones del SAF es por efecto tóxico directo del alcohol, a causa de deficiencias nutricionales asociadas o si se debe a posibles trastornos de la perfusión sanguínea intrauterina.
Las manifestaciones del SAF se han agrupado en 4 categorías (89): 1) disfunción y malformaciones del SNC (retardo mental, irritabilidad, temblor, convulsiones, hipotonía, síntomas cerebelosos), 2) retardo pre y postnatal del crecimiento, 3) anomalías cráneo-faciales características (hipoplasia de la cara, fisuras parpebrales cortas, pliegues epicánticos, puente nasal bajo con nariz corta y labio superior hipoplásico) y 4) malformaciones orgánicas y en las extremidades.
Pese a la alta incidencia del SAF, los estudios neuropatológicos detallados son relativamente escasos (90,91,92,93). Las lesiones más constantes son: malformaciones cerebrales y cerebelosas de gravedad variable (microcefalia, hidrocefalia, hipoplasia cerebelosa, porencefalia, arrinencefalia con agenesia del cuerpo calloso, siringomielia, malformación de Dandy-Walker, hidranencefalia), reducción de la sustancia blanca cerebral, heterotopias neurogliales periventriculares y leptomeníngeas. Son frecuentes las anomalías oculares. Estudios cuantitativos realizados en corteza cerebral con el método de Golgi revelan en las células piramidales hipoplasia dendrítica y disminución significativa del número de espinas con alteraciones de su morfología (92).
En animales de experimentación se ha demostrado el desarrollo de numerosas alteraciones que confirman el efecto nocivo del alcohol sobre el SN en desarrollo y que varían según la fase en que se aplica el tóxico. Así, se pueden producir malformaciones cerebrales, alteraciones de la emigración neuronal y heterotopias neurogliales, retardo de la maduración cerebelosa, trastornos de la gliogénesis, alteraciones oculares, etc. (84). Tambien se ha encontrado disminución de la expansión dendrítica en neuronas corticales (94). Parece que algunos cambios revierten en el período postnatal (95).
BIBLIOGRAFÍA
Graham DI, Thomas A, Gennarelli A. Trauma. En: Greenfield’s Neuropathology, ed por DI Graham y PL Lantos. London: Oxford University Press 1997, V.I: 197-262.
Kalimo H, Kaste M, Haltia M. Vascular diseases. En: Greenfield’s Neuropathology, ed por DI Graham y PL Lantos. London: Oxford University Press 1997, V.I: 315-96.
Cavanagh JB. Toxic and deficiency disorders. En: Symmer’s Systemic Pathology, V.4: Nervous System, Muscle and Eyes, ed por RO Weller. Edinburgh: Churchill Linigstone, 1990: 244-308.
Verity MA. Toxic disorders. En: Greenfield’s Neuropathology, ed por DI Graham y PL Lantos. London: Oxford University Press 1997, V.I: 755-811.
Cravioto H, Korein J, Silberman J. Wernicke’s encephalopathy: A clinical and pathological study of 28 autopsied cases. Arch Neurol 1961; 4: 510-19.
Victor M. The Wernicke-Korsakoff syndrome. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology Vol 28. Amsterdam: North-Holland Publishing Co 1976: 243-70.
Harper C. The incidence of Wernicke’s encephalopathy in Australia -A neuropathological study of 131 cases. J Neurol Neurosurg Psichiatry 1983; 46: 593-8.
Torvik A. Topographic distribution and severity of brain lesions in Wernicke’s encephalopathy. Clin Neuropathol 1987; 6: 25-9.
Harper C, Butterworth R. Nutritional and metabolic disorders. In: Graham DI, Lantos PL, eds. Greenfield’s Neuropathology. London: Edward Arnold 1997: 601-655.
Charness ME, De La Paz RL. Mammillary body atrophy in Wernicke’s encephalopathy: antemortem identification using magnetic resonance imaging. Ann Neurol 1987; 22: 595-600.
Naidoo DP, Bramdev A, Cooper K. Autopsy prevalence of Wernicke’s encephalopathy in alcohol-related disease. S Afr Med J 1996; 86: 1110-2.
Pena CE. Wernicke’s encephalopathy: report of seven cases with severe nerve cell changes in the mamillary bodies. Am J Clin Pathol 1969; 51: 603-9.
Byrne C, Halliday G, Ellis J, Harper C. Thalamic vacuolation in acute Wernicke’s encephalopathy. Metab Brain Dis 1993; 8: 107-13.
Mair WGP, Warington GK, Weiskrantz L. Memory disorder in Korsakoff’s psychosis. Brain 1979; 102: 749-83.
Mayes AR, Meudell PR, Mann D, Pickering A. Locations of the lesions in Korsakoff syndrome: neuropathological data on two patients. Cortex 1988; 24: 367-88.
Halliday G, Cullen K, Harding A. Neuropathological correlates of memory dysfunction in the Wernicke-Korsakoff syndrome. Alcohol Alcoholism 1994; 2(Suppl): 247-53.
Torvik A. Two types of brain lesions in Wernicke’s encephalopathy. Neuropathol Appl Neurobiol 1985; 11: 179-90.
Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s disease. Acta Neuropathol (Berl) 1983; 61: 101-8.
Halliday G, Ellis J, Harper C. The locus coeruleus and memory: a study of chronic alcoholis with and without the memory impairment of Korsakoff’s psychosis. Brain Res 1992; 598: 33-7.
Halliday G, Ellis J, Heard R, Caine D, Harper C. Brainstem serotonergic neurons in chronic alcoholics with and without the memory impairment of Korsakoff’s psychosis. J Neuropathol Exp Neurol 1993; 52: 567-79.
Ishii N, Nishihara Y. Pellagra among chronic alcoholics: Clinical and pathological study of 20 necropsy cases. J Neurol Neurosurg Psychiat 1981; 44: 209-15.
Hauw JJ, De Baecque C, Hausser-Hauw C, Serdaru M. Chromatolysis in alcoholic encephalopathies. Pellagra-like changes in 22 cases. Brain 1988; 111: 843-57.
Ishii N, Nishihara Y. Pellagra encephalopathy among tuberculous patients: its relation to isoniazid therapy. J Neurol Neurosurg Psychiatry 1985; 48: 628-34.
Serdaru M, Hausser-Hauw C, Laplane D et al. The clinical spectrum of alcoholic pellagra encephalopathy. A retrospective analysis of 22 cases studied pathologically. Brain 1988; 111: 829-42.
Leigh D. Pellagra and the nutritional neuropathies: a neuropathological review. J Ment. Sci 1952; 98:130-142.
Charness ME, Simon RP, Greenberg DA. Ethanol and the nervous system. N Engl J Med 1989; 321: 442-54.
Kril JJ., Halliday GM, Svoboda MD, Cartwright H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience 1997; 79: 983-98.
Harper CG, Blumberg PC. Brain weights in alcoholics. J Neurol Neurosurg Psychiat 1982; 45: 838-40.
Torvik A, Lindboe CF, Rodge S. Brain lesions in alcoholics. A neuropathological study with clinical correlations. J Neurol Sci 1982; 56: 233-48.
Harper C, Kril JJ. Brain atrophy in chronic alcoholic patients: a quantitative pathological study. J Neurol Neurosurg Psychiat 1985; 48: 211-7.
De La Monte SM Disproportionate atrophy of cerebral white matter in chronic alcoholics. Arch Neurol 1988; 45: 990-2.
Jernigan TL, Schafer K, Butters N, Cermak LS. Magnetic resonance imaging of alcoholic Korsakoff’s patients. Neuropsycho-pharmacol 1991; 4: 175-86.
Kril JJ, Harper CG. Neuronal counts from four cortical regions of alcoholic brains. Acta Neuropathol (Berl) 1989; 79: 200-4.
Jensen GB, Pakkenberg B. Do alcoholics drink their neurons away? Lancet 1993; 342: 1201-4.
Löken AC. Ethyl Alcohol. En: Pathology of the Nervous System, V.2, ed. por J Minckler. New York, McGraw-Hill, 1971: 1671-8.
Castaigne P, Buge A, Cambier J, Escourolle R, Rancurel G. La maladie de Marchiafava-Bignami: Étude anatomoclinique de dix observations. Rev Neurol (Paris) 1971: 125: 179-96.
Naeije R, Franken L, Jacobovitz D, Flament-Durand J. Morel’s laminar sclerosis. Eur Neurol 1978; 17: 155-9.
Regadera JF, Enriquez R, Morales M, Gutierrez M, de Lorenzo A, Cambronero JA. Asociación de enfermedad de Marchiafava-Bignami con mielinolisis pontina central y esclerosis laminar de Morel: presentación de un caso. Med Clin (Barc) 1984; 82: 117-20.
Okeda R, Kitano M, Sawabe M, Yamada I, Yamada M. Distribution of demyelinating lesions in pontine and extrapontine myelinolysis—three autopsy cases including one case devoid of central pontine myelinolysis. Acta Neuropathol (Berl), 1986; 69: 259-66.
Victor M, Adams RD, Mancall EL. A restricted form of cerebellar cortical degeneration occurring in alcoholic patients. Arch Neurol 1959; 71: 579-688.
Torvik A, Torp S. The prevalence of alcoholic cerebellar atrophy. A morphometric and histological study of autopsy material. J Neurol Sci 1986; 75: 43-51.
Phillips SC, Harper CG, Kril JJ. A quantitative histological study of the cerebellar vermis in alcoholic patients. Brain 1987; 110: 301-14.
Phillips SC, Harper CG, Kril JJ. The contribution of Wernicke’s encephalopathy to alcohol-related cerebellar damage. Drug Alcohol Rev 1990; 9: 53-60.
Ferrer I, Fábregues I, Pineda M, Gracia I, Ribalta T. A Golgi study of cerebellar atrophy in human chronic alcoholism. Neuropathol Appl Neurobiol 1984; 10: 245-53.
Marchiafava E, Bignami A. Sopra un alterazione del corpo callosa osservata in sogetti alcoolista. Riv Patol Nerv Ment 1903; 8: 544-9.
Koeppen AH, Barron KD. Marchiafava-Bignami disease. Neurology 1978; 28: 290-4.
Leong ASV. Marchiafava-Bignami disease in a non-alcoholic Indian male. Pathology 1979; 11: 241-9.
Ghatak NR, Hadfield MG, Rosenblum WI. Association of central pontine myelinolysis and Marchiafava-Bignami disease. Neurology 1978; 28: 1295-8.
Kawamura M, Shiota J, Yagishita T, Hirayama K. Marchiafava-Bignami disease: computed tomographic scan and magnetic resonance imaging. Ann Neurol 1985; 18: 103-4.
Tobita M, Mochizuki H, Takahashi S, Onodera H, Itoyama Y, Iwasaki Y. A case of Marchiafava-Bignami disease with complete recovery: sequential imaging documenting improvement of callosal lesions. Tohoku J Exp Med, 1997; 182: 175-9.
Izquierdo G, Quesada MA, Chacon J, Martel J. Neuroradiologic abnormalities in Marchiafava-Bignami disease of benign evolution. Eur J Radiol, 1992; 15: 71-4.
Canaple S, Rosa A, Mizon JP. Maladie de Marchiafava-Bignami: disconnexion interhemispherique, evolution favorable. Aspect neuroradiologique. Rev Neurol (Paris) 1992; 4: 638-40.
Ironside R, Bosanquet FD, McMenemey WH. Central demyelination of the corpus callosum (Marchiafava-Bignami disease) with report of a second case in Great Britain. Brain 1961; 84: 212-30.
Shiota JI, Nakano I, Kawamura M, Hirayama K. An autopsy case of Marchiafava-Bignami disease with peculiar chronological CT changes in the corpus callosum: neuroradiopathological correlations. J Neurol Sci 1996; 136: 90-3.
Pfefferbaum A, Lim KO, Desmond JE, Sullivan EV. Thinning of the corpus callosum in older alcoholic men: a magnetic resonance imaging study. Alcohol Clin Exp Res 1996; 20: 752-7.
Adams RA, Victor M, Mancall EL. Central pontine myelinolysis: A hitherto undescribed disease occurring in alcoholic and malnourished patients. Arch Neurol Psychiatry 1959; 81: 154-72.
Wright DG, Laureno R, Victor M. Pontine and extrapontine myelinolysis. Brain 1979; 102: 361-385.
Estol CJ, Faris AA, Martínez J, Ahdab-Barmada M. Central pontine myelinolysis after liver transplantation. Neurology 1989; 39: 493-8.
Thompson DS, Hutton JT, Stears JC, Sung JH, Norenberg MD. Computed tomography in the diagnosis of central and extrapontine myelinolysis. Arch Neurol 1981; 38: 243-6.
Brunner JE, Redmond JM, Haggar AM, Elias SB. Central pontine myelinolysis after rapid correction of hyponatremia: A magnetic resonance imaging study. Ann Neurol 1988; 23: 389-91.
Clifford DB, Gado MH, Levy BK. Osmotic demyelination syndrome: lack of pathologic and radiologic imaging correlation. Arch Neurol 1989; 46: 343-7.
Goebel HH, Zue PH. Central pontine myelinolysis. A clinical and pathological study of 10 cases. Brain 1972; 95: 495-504.
Gocht A, Lohler J. Changes in glial cell markers in recent and old demyelinated lesions in central pontine myelinolysis. Acta Neuropathol (Berl) 1990; 80: 46-58.
Forno LS, Rivera L. Central pontine myelinolysis. J Neuropathol Exp Neurol 1975; 34: 77.
Powers JM, McKeever PE. Central pontine myelinolysis. An ultrastructural and elemental study. J Neurol Sci 1976; 29: 65-81.
Gocht A, Colmant HJ. Central pontine and extrapontine myelinolysis: a report of 58 cases. Clin Neuropathol 1987; 6: 262-70.
McComb RD, Peiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hypoglycemia. Clin Neuropathol 1989; 8: 284-8.
Messert B, Orrison WM, Quaglieri CE. Central pontine myelinolysis: consideration on etiology, diagnosis, and treatment. Neurology 1979; 29: 2-60.
Norenberg MD, Leslie KO, Robertson AS. Association between rise in serum sodium and central pontine myelinolysis. Ann Neurol 1982; 11: 128-35.
Laureno R. Central pontine myelinolysis following rapid correction of hyponatremia. Ann Neurol 1983; 13: 232-42.
Sterns RH, Riggs JE, Schochet SS. Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med 1986; 314: 1535-42.
McKee AC, Winkleman MD, Banker BQ. Central pontine myelinolysis in severely burned patients. Relationship to serum hyperosmolality. Neurology 1988; 38: 1211-7.
Riggs JE, Schochet SS. Osmotic stress, osmotic myelinolysis, and oligodendrocyte topography. Arch Pathol Lab. Med 1989; 113: 1386-8.
Peeters A, Van de Wyngaert F, Van Lierde M, Sindic CJ, Laterre EC. Wernicke’s encephalopathy and central pontine myelinolysis induced by hyperemesis gravidarum. Acta Neurol Belg 1993; 93: 276-82.
Norenberg MD. A hypothesis of osmotic endothelial injury: a pathogenetic mechanism in central pontine myelinolysis. Arch Neurol 1983; 40: 66-9.
Denny-Brown D. The neurological aspects of thiamine deficiency. Fed Proc 1958; 17 (Suppl 2): 35-9.
Bischoff A. Die alkoholische Polyneuropathie: klinische, ultrastrukturelle und pathogenetische Aspekte. Dtsch Med Wochenschr 1971; 96: 317-22.
Walsh JC, McLeod JG. Alcoholic neuropathy. An electrophysiological and histological study. J Neurol Sci 1970; 18: 459-69.
Behse F, Buchtal F. Alcoholic neuropathy: clinical, electrophysiological, and biopsy findings. Ann Neurol 1977; 2: 95-110.
Said G, Landrieu P. Etude quantitative des fibres nerveuses isolées dans les polynévrites alcooliques. J Neurol Sci 1978; 35: 317-30.
Barter F, Tanner AR. Autonomic neuropathy in an alcoholic population. Postgrad Med J 1987; 63: 1033-36.
Johnson RH, Bobinson BJ. Mortality in alcoholics with autonomic neuropathy. J Neurol Neurosurg Psichiat 1988; 51: 476-80.
Sage JI, Van Uitert RL, Lepore FE. Alcoholic myelopathy without substantial liver disease. A syndrome of progressive dorsal and lateral column dysfunction. Arch Neurol 1984; 41: 999-1001.
Weller RO, Cumming WJK, Mahon M. Diseases of muscle. En: Greenfield’s Neuropathology, ed por DI Graham y PL Lantos. London: Oxford University Press 1997, V.2: 489-581.
Thomas PK, Landon DN, King RHM. Pathology of muscle diseases. En: Symmer’s Systemic Pathology, V.4: Nervous System, Muscle and Eyes, ed por RO Weller. Edinburgh: Churchill Linigstone, 1990: 604-65.
Seitelberger F. Neuropathological conditions related to neuroaxonal dystrophy. Acta Neuropathol (Berlin) 1871; (Suppl V): 17-29.
Yagishita S. Morphological investigation of axonal swellings and spheroids in various human diseases. Virchows Arch Path Anat 1978; 378: 181-97.
Jellinger K. Neuroaxonal dystrophy: Its natural history and related diseases. Progr Neuropathol 1973; 2: 129-80.
Clarren SK, Smith DW. The fetal alcohol syndrome: A review of the world literature. New Eng J Med 1978; 298: 1063-7.
Peiffer J, Majewski F, Fischbach H, Bierch JR, Volk B. Alcohol embryo- and fetopathy: neuropathology of 3 children and 3 fetus. J Neurol Sci 1979; 41: 125-37.
Wisniewski K, Dambska W, Sher JH, Qazi Q. A clinical neuropathology study of the fetal alcohol syndrome. Neuropadiatrics 1983; 14: 197-201.
Ferrer I, Galofre E. Dendritic spine abnormalities in fetal alcohol syndrome. Neuropediatrics 1987; 18: 161-3.
Coulter CL, Leech RW, Schaefer GB, Scheithauer BW, Brumback RA. Midline cerebral dysgenesis, dysfunction of the hypothalamic-pituitary axis, and fetal alcohol effects. Arch Neurol 1993; 50: 771-5.
Fábregues I, Ferrer I, Gairi JM, Cahuana A, Giner P. Effects of prenatal exposure to ethanol on the maturation of the pyramidal neurons in the cerebral cortex of the guinea pig: A quanttitative Golgi study. Neuropathol Appl Neurobiol 1985; 11: 291-8.
López-Tejero D, Ferrer I, Llobera M, Herrera E. Effects of prenatal ethanol exposure on physical growth, sensory reflex maturation and brain development on the rat. Neuropathol Appl Neurobiol 1986; 12: 251-60.
![]()