
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
Ruth Orellana Fernández, Jaume Almirall Daly1, Fco. Javier Andreu Navarro
Servicio de Anatomía Patológica y Unidad de Nefrología1. Corporació Sanitària Parc Taulí. Sabadell (Barcelona)
INTRODUCCIÓN-GENERALIDADES
Está demostrado que los virus pueden actuar como agente etiológico en enfermedades renales en animales y en modelos experimentales. La infección vírica provocaría daño renal por diferentes mecanismos. Los mecanismos patogénicos implicados comprenden desde 1) acción citopática directa por el virus (como es el caso de glomerulonefritis en el curso de infecciones congénitas por citomegalovirus); y 2) mecanismo autoinmunitario con la formación ya sea directa o inducida de inmunocomplejos y posterior depósito a distintos niveles del parénquima renal (como sería el caso de asociación de infección por virus B de la hepatitis y glomerulonefritis membranosa).
En humanos, la evidencia de glomerulonefritis de etiología vírica es indirecta. La asociación temporal puede proporcionar una evidencia altamente sugestiva de relación causal entre glomerulonefritis e infección viral aguda (documentada a partir de criterios clínicos, determinación de carga viral o respuesta serológica); sin embargo, este tipo de evidencia continúa siendo circunstancial. La ubicuidad de muchos de los agentes víricos implicados restaría especificidad a las determinaciones serológicas y la demostración ultraestructural de partículas víricas o virus-like en glomérulos o parénquima en general, tampoco debería considerarse evidencia definitiva de etiología vírica. Por ejemplo, aunque la nefritis lúpica no se cree causada directamente por virus, con frecuencia se observan partículas virus-like en el estudio ultraestructural o antígenos de oncornavirus a nivel glomerular (1). Por otro lado, en muestras renales de pacientes con lupus o glomerulonefritis por inmunocomplejos idiopática se pueden determinar anticuerpos anti-sarampion o anti-virus Epstein Barr, coincidiendo con títulos elevados en suero de anticuerpos frente a los mencionados virus y no significando necesariamente una implicación etiológica.
Se han descrito cuadros de glomerulonefritis proliferativa endocapilar con o sin fenómenos de arteritis asociados, y en los que serológicamente se había descartado una glomerulonefritis post estreptocócica concomitante, con niveles de ASLO normales, en el curso de infección por virus de la varicela, virus del sarampión, virus de la parotiditis, virus Epstein Barr (VEB) e influenza y con un curso evolutivo similar a las glomerulonefritis agudas clásicas (2). Otras formas de afectación glomerular incluye la glomerulonefritis focal y segmentaria (glomerulopatía colapsante) en infecciones por parvovirus B19 (3), en pacientes susceptibles. Se ha comprobado una alta incidencia de serología positiva para virus Coxackie B en niños con síndrome hemolítico-urémico, en los que también se ha detectado antígeno viral en el parénquima renal por técnica de inmunofluorescencia, sin concluirse una implicación etiológica definitiva (2). Otros ejemplos de afectación renal en el curso de infecciones víricas incluyen nefropatías túbulointersticiales (1) en pacientes con infección por citomegalovirus, poliomavirus, virus Epstein Barr, hantavirus, adenovirus, ecovirus, etc., o casos aislados de mioglobinuria, coagulación intravascular y necrosis tubular aguda en el curso de infección por virus influenza (1,4).
En resumen, a pesar de haberse descrito diferentes nefropatías en el curso de infecciones víricas, es difícil atribuir un papel etiológico directo del virus y diferenciarlo de una mera asociación circunstancial. No obstante, hay diversos escenarios clínicopatológicos en los que la implicación vírica está bien establecida y en los que el diagnóstico patológico adquiere una trascendencia decisiva a la hora de plantear las diferentes alternativas terapéuticas. En este sentido, destacamos las siguientes situaciones:
1. Virus y trasplante renal.
2. Nefropatía asociada a infección por HIV (HIVAN).
3. Virus C de la hepatitis y nefropatía.
VIRUS Y TRASPLANTE RENAL
Las infecciones víricas son una causa importante de morbilidad y mortalidad postrasplante (5). Por un lado, el estado de inmunodepresión del paciente trasplantado, junto a la indicación de agentes inmunosupresores cada vez más potentes son responsables, en parte, del aumento de incidencia de algunas de estas infecciones víricas. Además, muchas de estas infecciones comprometen el funcionalismo del injerto, con una semiología similar a la del rechazo. En esta situación concreta, el diagnóstico diferencial es trascendente, dada la diferente orientación terapéutica; y comprende criterios clínicos, serológicos, biopsia renal y confirmación con técnicas de inmunohistoquímica, estudio ultraestructural y en algunos casos técnicas de biología molecular, con la reciente incorporación de tests más rápidos y fiables.
Se revisan los aspectos clínicos y patológicos de las infecciones víricas más frecuentes en el curso del trasplante renal, con énfasis exclusivo en aquellas infecciones con repercusión funcional del injerto. En este sentido, aunque se describen infecciones por herpesvirus 3 (virus varicela-zoster, responsable ocasionalmente de cuadros severos de afectación visceral post-trasplante), herpesvirus (6,7) (responsables de cuadros exantemáticos), herpesvirus 8 (agente etiológico del sarcoma de Kaposi) y virus respiratorios (influenza, parainfluenza, virus respiratorio sincitial y adenovirus) responsables de cuadros de neumonitis y cistitis hemorrágica en trasplantados, no se ha demostrado su implicación lesional a nivel renal. Por otro lado, igual que en pacientes no trasplantados, el virus C de la hepatitis se ha relacionado con glomerulonefritis mediadas por inmunocomplejos, característicamente con patrón lesional membranoproliferativo, sin poder precisar su repercusión en la evolución funcional del injerto y la supervivencia del paciente trasplantado (6).
1. HERPES VIRUS
Los herpes virus son virus DNA que de forma característica permanecen en estado latente después de la infección primaria y pueden manifestarse varios años después de la exposición inicial. Se conocen 8 herpesvirus patógenos en humanos (HHV1-8). El diagnóstico serológico sólo es útil como marcador de exposición previa.
HHV-1 y 2: Herpes simple 1 y 2
En 50-66% de trasplantados seropositivos, se pueden detectar viremias entre 5 y 14 días después del trasplante, aunque sólo un 15-45% de éstos presentará sintomatología en forma de úlceras o vesículas (7).
Son responsables de meningoencefalitis y más raramente de hepatitis potencialmente severas. Sólo se conoce un caso de nefritis intersticial con fracaso del injerto atribuido a infección por HSV-1 (8).
El diagnóstico definitivo se basa en el cultivo de líquido contenido en las vesículas, frotis de mucosas, líquido cefalorraquídeo (LCR) u orina. Para el diagnóstico se utiliza también reacción en cadena de la polimerasa (PCR), principalmente en líquido cefalorraquídeo.
HHV-4: Virus Epstein-Barr
El virus de Epstein Barr (VEB) se ha implicado en diversas situaciones patológicas en el paciente trasplantado, aunque característicamente se ha relacionado con el síndrome linfoproliferativo postrasplante (SLPT) (9), que se produce en el 2% de los receptores de trasplante renal (10). El SLPT resultaría de una proliferación incontrolada de células B transformadas por el VEB y favorecida por el estado de inmunosupresión. Clínicamente puede presentarse de forma variada, desde un síndrome vírico (incluyendo la mononucleosis infecciosa) hasta un linfoma de células B agresivo, con situaciones clínicopatológicas intermedias (11). En el registro de Cincinnati, el SLPT representa el 22% de los tumores postrasplante (12). Las formas más agresivas de SLPT se presentan en pacientes adultos y de forma tardía, varios años después del trasplante, como linfomas agresivos con predominante afectación extranodal. La afectación extranodal se produce en el 70% de los pacientes y la localización más frecuente es el sistema nervioso central (SNC), seguido del órgano trasplantado (20% y en un tercio de éstos como afectación exclusiva) y el tracto gastrointestinal. Se han descrito casos de afectación multiorgánica (5).
En casos con afectación del injerto, se plantea el diagnóstico diferencial con una forma severa de rechazo agudo, pues la presencia de venulitis, tubulitis e infiltración de tejidos perihiliares pueden aparecer en ambas patologías. La presencia de infiltrados linfoides expansivos y destructivos con focos de necrosis, junto a la marcada atipia nuclear de los elementos proliferados, sugiere SLPT (13) (fig. 1a). El estudio inmunohistoquímico en el SLPT detecta linfocitos transformados por el VEB, predominantemente de inmunofenotipo B (CD-20 positivos), a diferencia del inmunofenotipo T predominante en el rechazo agudo (fig. 1b). La demostración del RNA del VEB (EBER) por hibridación in situ se consigue en el 90% de los casos de SLPT, mientras que con técnicas de inmunohistoquímica se detecta la presencia de proteínas latentes de membrana (LPM-1) y EBNA, en aproximadamente el 50% de las lesiones (fig. 1c). El diagnóstico diferencial con el rechazo es decisivo, dado el diferente manejo terapéutico.
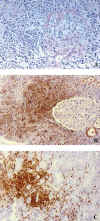
Fig. 1. Virus
Epstein Barr: (a) Infiltración por linfocitos en diferentes estadios de
diferenciación (PAS x 450); (b) linfocitos predominantemente de inmunofenotipo
CD20+ (PO x 250) y (c) linfocitos mostrando positividad para proteína latente
de membrana (LPM-1), (PO x 450).
HHV-5: Citomegalovirus
Los rangos de seropositividad para el citomegalovirus (CMV) entre diferentes poblaciones oscilan entre 40-80%, con un amplio espectro de cepas genéticamente diferentes. La seropositividad para CMV no implica por ello inmunidad y la reinfección con cepas diferentes puede causar enfermedad sintomática (5).
En el paciente trasplantado, la enfermedad clínicamente aparente se puede producir desde a) una cepa latente en el receptor o b) desde una infección primaria o como superinfección, originadas en el órgano trasplantado o c) desde la sangre de un donante seropositivo. La terapia antilinfocítica con OKT3 está asociada con un mayor riesgo de infección primaria sintomática y con riesgo de 2 a 10 veces de reactivación en pacientes seropositivos (14). Los receptores seronegativos de un injerto con donante seropositivo, muestran seroconversión hacia los 6 meses del trasplante. Sin tratamiento profiláctico, un 50% desarrollarán enfermedad sintomática (caracterizada por fiebre, astenia, elevación de transaminasas, linfocitosis atípica) y puede acompañarse de afectación funcional del injerto, signos de afectación pulmonar, gastrointestinal, etc. Un 10% de pacientes inicialmente seropositivos presentarán infección sintomática por CMV, independientemente del estatus serológico del donante (15).
El injerto es un órgano diana frecuente, tanto de infecciones subclínicas como activas. En infecciones activas, se identifican inclusiones características citomegálicas en el epitelio tubular, endotelio de glomérulos y vasos peritubulares y en elementos inflamatorios. La infección puede asociarse a disfunción del injerto en ausencia de inflamación o, más frecuentemente, en el contexto de glomerulitis o nefritis túbulointersticial similar a la observada en el rechazo agudo. Por otro lado, la infección por CMV puede predisponer a la aparición de rechazo agudo (16), por mecanismos en los que se implica la probable disregulación de antígenos HLA tipo II. Por último, la infección por CMV se ha identificado como factor de riesgo para el desarrollo de rechazo crónico, aunque restringida a pacientes con infección por CMV y rechazo agudo previo (17).
El diagnóstico se basa en la detección del virus en sangre periférica o por hallazgos diagnósticos en el estudio histológico, incluyendo positividad inmunohistoquímica para antígenos virales (18) (figs. 2a y 2b).
![]()
Fig. 2. Citomegalovirus:
(a) Típicas inclusiones intranucleares por citomegalovirus en el epitelio
tubular y en el epitelio parietal de la cápsula de Bowman (H&E x 450). (b)
Positividad para el antígeno viral (PO x 650).
2. POLIOMAVIRUS
Los virus del polioma son un grupo de virus DNA de doble hélice. Son ubicuos y los estudios serológicos han demostrado que alrededor del 100% de la población sufre infección durante la infancia, normalmente asintomática (19).
Esta familia de virus incluye virus JC y BK, el primero es más conocido como agente causal de la leucoencefalopatía multifocal progresiva; el BK-virus después de una infección primaria queda latente en el epitelio tubular renal (20). La reactivación del virus se puede producir en el curso de un trasplante renal, como resultado del estado de inmunosupresión, y puede ser detectado en el 20% de los pacientes (21). El pico de incidencia de infección por virus del polioma se produce durante los primeros seis meses postrasplante, aunque la infección puede persistir durante meses o incluso años. El virus muestra tropismo por el tracto urinario, incluyendo epitelio de vejiga urinaria, uréter y túbulos renales. La reactivación sólo se manifiesta clínicamente en el 1-2% de los pacientes, en forma de cistitis hemorrágica, estenosis ureteral y nefritis intersticial.
Histológicamente, la infección provoca una nefritis tubulointersticial que puede similar un rechazo agudo (22). Es frecuente la presencia de células plasmáticas en el infiltrado intersticial y mayor grado de afectación tubular que el observado en tubulitis de rechazo, con el que debe establecerse el diagnóstico diferencial. El núcleo de las células epiteliales aparece agrandado, con cromatina de aspecto deslustrado e inclusiones víricas basófilas en vidrio esmerilado o rodeadas por un halo, casi siempre incompleto (figs. 3a y 3b). La presencia de infección por virus del polioma se confirma con la tinción inmunohistoquímica para antígeno SV40, que presenta reacción cruzada con el virus BK, técnicas de hibridación in situ y estudio ultraestructural (fig. 3c). Las células infectadas por virus del polioma pueden detectarse mediante estudio citológico de orina, considerado un método sensible y no invasivo para la detección de infección con significación clínica (23). La infección por virus del polioma responde a la disminución de terapia inmunosupresiva.
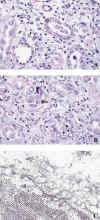
Fig. 3. Virus
BK: (a) Inclusiones basófilas en vidrio esmerilado (H&E x 450); (b)
Inclusiones víricas basófilas con patrón difuso o vesiculares rodeadas por un
halo incompleto (H&E x 450) y, (c) viriones del polipoma BK (ME x 26.000).
NEFROPATÍA ASOCIADA A HIV
Aunque la mayoría de las alteraciones renales en el paciente con infección por HIV son debidas a procesos intercurrentes, sepsis de diversa etiología, deshidratación, nefrotoxicidad por fármacos, etc. a partir de 1984, se describe una nefropatía específica asociada a HIV (HIVAN), reconocida como entidad clínicopatologica (24). Un 10% de pacientes infectados por HIV desarrollarían HIVAN. Epidemiológicamente, la HIVAN se observa predominantemente en individuos de raza negra, por lo que en nuestro medio es poco prevalente. Esta afectación renal acostumbra a presentarse clínicamente como síndrome nefrótico con pérdida progresiva de la función renal, frecuentemente en forma de insuficiencia renal rápidamente progresiva, requiriendo tratamiento substitutivo con diálisis, en pocos meses (25). Otros datos clínicos característicos de esta entidad (y no propios de la nefropatía asociada a consumo de heroína por vía parenteral, entidad con la que inicialmente se había relacionado patogénicamente) incluirían la ausencia de edema franco a pesar de la proteinuria de rango nefrótico habitual, la ausencia de hipertensión arterial importante y el tamaño renal conservado en el estudio ecográfico (26).
El aspecto histológico de las lesiones renales es característico (27). A nivel glomerular se observa (fig. 4a) una lesión de glomerulosclerosis focal y segmentaria (GFS) con un porcentaje variable de glomérulos esclerosados globalmente (también conocida como glomerulosclerosis colapsante). Otros hallazgos glomerulares incluyen podocitos de aspecto hipertrófico, con citoplasmas ocasionalmente vacuolizados o espumosos y cambios isquémicos en la pared capilar glomerular. En estadios iniciales puede observarse ensanchamiento de matriz mesangial. En el intersticio se observa fibrosis, de severidad desproporcionada con la afectación glomerular y compromiso inflamatorio de células redondas y de distribución parcheada. Otra característica histológica es la dilatación ectásica de los túbulos renales, con abundantes cilindros hialinos. Se observa depósitos de IgM y fracciones del complemento en las zonas de esclerosis glomerular y fijación mesangial en menor grado. Ultraestructuralmente, se han detectado inclusiones túbuloreticulares en células endoteliales de capilares glomerulares y peritubulares y en elementos inflamatorios intersticiales (fig. 4b).
![]()
Fig. 4. HIV:
(a) Lesión glomerular de esclerosis focal y segmentaria (Tricrómico de Masson
x 600) y, (b) inclusiones tuboloreticulares en endotelio capilar glomerular y en
el citoplasma de una célula mononuclear circulante (ME x 8.500).
Se han descrito otras formas de afectación glomerular en pacientes con infección por HIV, mediadas o no por inmunocomplejos (28) (nefropatías IgA (29), hiperplasia mesangial sin GFS, glomerulonefritis membranosa asociada a virus B de la hepatitis concomitante (30), glomerulopatía por cambios mínimos, glomerulonefritis membranoproliferativa, glomerulonefritis proliferativa difusa lupus-like). La relación patogénica de estas lesiones con la infección por HIV no está establecida (31).
En la actualidad, a pesar de la introducción de agentes antiretrovirales, no se ha demostrado la eficacia de ninguna pauta terapéutica en la evolución de la HIVAN. Muchos de estos pacientes evolucionan a insuficiencia renal terminal, con necesidad de tratamiento dialítico, con alta morbi-mortalidad asociada.
VIRUS C DE LA HEPATITIS Y NEFROPATÍA
Aparte de la glomerulonefritis membranosa (y más raramente glomerulonefritis membranoproliferativa, glomerulonefritis proliferativa mensangial IgG, IgM o nefropatía por depósitos de IgA), asociada de forma clásica a virus B de la Hepatitis, en los últimos años, el virus C de la Hepatitis ha tomado protagonismo en cuanto a su relación con la nefropatología.
Desde el descubrimiento hace una década de la existencia del VHC se ha producido un aumento progresivo de información que relaciona dicha infección con la existencia de múltiples alteraciones a nivel extrahepático. A nivel renal se han descrito de forma aislada múltiples patrones de lesión glomerular (glomerulosclerosis focal y segmentaria, nefropatía por depósitos de IgA, glomerulopatía inmunotactoide, etc.). Sin embargo, el análisis riguroso de la relación estadística de estas asociaciones han sido cuestionada, dada la alta prevalencia de seropositivos en la población general (2-3%). Actualmente existen tres procesos renales claramente relacionados con la infección del VHC: crioglobulinemia mixta, glomerulonefritis membranoproliferativa y glomerulonefritis membranosa (aunque cuestionada por algunos autores).
Crioglobulinemia mixta
Las crioglobulinas (CG) son inmunoglobulinas (Ig) con la propiedad de precipitar de forma reversible con el frío. Desde 1974 se mantiene la clasificación de Brouet en tres tipos: Tipo I, CG formadas por una Ig monoclonal única; Tipo II, constituida por una Ig policlonal (generalmente IgG) y una Ig monoclonal (tipo IgM) y la Tipo III constituida por dos Ig policlonales.
La crioglobulinemia mixta (CM) tipo II se caracteriza clínicamente por una vasculitis sistémica que cursa con púrpura, artralgias, astenia y posibilidad de afectación multiorgánica, entre ellas la afectación renal. Estas manifestaciones son debidas al depósito de CG que pueden ocasionar oclusión vascular con isquemia o infarto o bien por el desarrollo de un proceso inflamatorio de la pared vascular.
La asociación entre la infección por VHC y CM tipo II se ha consolidado en los últimos años (32,33). Algunos autores han sugerido que dicha infección estaría presente en la mayoría de casos (>80%) de CM tipo II, hasta entonces denominada «esencial» (34), por lo que dicho término casi debería desaparecer. Sin embargo la prevalencia y significado clínico de la CG que acompaña a la infección por el VHC no está totalmente esclarecida. Tampoco se conoce con exactitud con qué frecuencia se presentan manifestaciones clínicas extrahepáticas en ausencia de CG o cómo incidiría el tratamiento antiviral sobre estas manifestaciones clínicas. La infección por el VHC puede estar involucrada también en un porcentaje no despreciable de casos de CM tipo III, aunque éstos son clínicamente menos activos, en especial en cuanto a la afectación renal se refiere.
Las manifestaciones clínicas consisten en la existencia de hematuria, proteinuria (a menudo de rango nefrótico), hipertensión arterial y grados variables de insuficiencia renal, siendo el curso evolutivo muy variable. Las determinaciones serológicas confirman la presencia de crioglobulinas séricas y la disminución de fracciones del complemento.
La expresión morfológica característica a nivel glomerular (también llamada glomerulonefritis crioglobulinémica), corresponde a un patrón lesional membranoproliferativo, especialmente proliferativo con abundancia de células monocitarias y presencia ocasional de trombos intracapilares eosinofílicos, PAS positivos, constituidos por la CG (fig. 5a). La interposición de estos elementos entre la membrana basal y las células endoteliales condiciona la formación de nueva membrana basal, que se traduce en la presencia de «dobles contornos» con las técnicas argénticas. En ocasiones puede demostrarse la existencia de vasculitis afectando a estructuras vasculares extraglomerulares de distinto calibre (30% de casos). El examen mediante inmunoflorescencia directa revela un patrón de fijación mesangial y parietal florido para IgM, IgG y C3. Esta fijación es especialmente destacable en las zonas en que existen trombos intraluminales (fig. 5b). A nivel ultraestructural destacan la presencia de depósitos electrodensos de disposición subendotelial, mesangial y con menos frecuencia intramembranosos, y la existencia de «fingerprint», estructuras microtubulares de características superponibles al crioprecipitado obtenido de sangre periférica.
![]()
Fig. 5. Virus
C: (a) Glomerulonefritis crioglobulinémica (Tricrómico de Masson x 600) y, (b)
depósitos preferentemente intraluminares de IgM (IF x 450).
Glomerulonefritis membranoproliferativa (GNMP)
En diversos estudios se ha constatado la relación entre la infección por el VHC y la GNMP en ausencia de CG (35). Esta asociación ha sido cuestionada por diversos autores, dada la ausencia de información sobre la determinación de CG en algunos artículos y la dificultad metodológica para la determinación de las CG. Sin embargo, una revisión reciente destaca la existencia de una prevalencia del 15% de infección por el VHC (por encima del 2-3% de la seropositividad en la población general) entre todos los casos diagnosticados de GNMP, sugierendo una relación patogénica. Estos casos con CG negativas cursan de forma característica con manifestaciones sistémicas poco frecuentes (36).
Glomerulonefritis membranosa (GM)
La GM es otro patrón histológico que ha sido relacionado con la infección por el VHC (37). Aunque algunos autores defienden esta posibilidad, la evidencia en este sentido es mucho más débil. Un metaanálisis reuniendo la información disponible con un total de 291 casos de GM (36), encuentra una incidencia de infección por el VHC de 5,4% (no muy superior a la existente en la población general). En estos pacientes la determinación serológica de fracciones del complemento acostumbra a ser normal y la determinación de CG negativa.
AGRADECIMIENTOS
Las figuras: 1(a,b y c), 3(a y b), 4(a y b) y 5 (a y b) han sido proporcionadas por F. Mampaso (H. Univ. Ramón y Cajal, Madrid); 2(b) por J. Blanco (H. Clin.San Carlos , Madrid) y, 3(c) por I. García (H. Univ.Carlos Haya, Málaga).
BIBLIOGRAFÍA
Ronco P, Verroust P, Morel-Maroger L. Viruses and glomerulonephritis. Nephron 1982; 31: 97-102.
Gallo GR, Neugarten J, Baldwin DS. Glomerulonephritis associated with systemic bacterial and viral infections. En: Tisher CC, Brenner BM (Ed.). Renal pathology with clinical and functional correlations. J.B. Lippincot, Philadelphia 1989; 548-74.
Moudgil A, Nast C, Bagga A y cols. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int 2001; 59: 2126-33.
Morgasen JL. Myoglobinuria adn renal failure associated with influenza. Ann Intern. Med 1974; 80: 362-3.
Fishman JA, Rubin RH. Infection in organ-transplant recipients. N Engl J Med 1998; 338: 1741-51.
Cruzado JM, Torras J, Gil-Vernet S y Griñó JM. Glomerulonephritis associated with hepatitis C virus infection after renal transplantation. Nephrol Dial Transplant 2000; 15(Suppl 8): 65-7.
Greenberg MS, Friedman H, Cohen SG, Oh SH, Laster L, Starr S. A comparative study of herpes simplex infections in renal transplant and leukemic patients. J Infect Dis 1987; 156: 280-7.
Smith SR, Butterly DW, Alexander BD, Greenberg A. Viral infections after renal transplantation. Am J Kidney Dis 2001; 37 (4): 659-76.
Cohen JI. Epstein-Barr virus infection. N Engl J Med 2000; 343: 481-92.
Boubenider S, Hiesse C, Goupy C, Kriaa F, Marchand S, Charpentier B. Incidence and consequences of post-transplantation lymphoproliferative disorders. J Nephrol 1997; 10: 136-45.
Hanto DW. Classification of Epstein-Barr virus-associated pottransplant lymphoproliferative diseases: Implications for understanding their pathogenesis and developing rational treatment strategies. Annu Rev Med 1995; 46: 381-94.
Penn I. Immunosupresion: A contributory factor in lymphoma formation. Clin Transpl 1992; 6: 214-9.
Randhawa PS, Demetris AJ, Piertzak b, Nalesnik MA. Histopathology of renal pst-transplant proliferations: comparison with rejection using the Banff schema. Am J Kidney Dis 1996; 28: 578-84.
Hibberd PL, Tolkoff-Rubin NE, Cosimi AB and cols. Symptomatic cytomegalovirus disease in the cytomegalovirus antibody seropositive renal transplant recipient treated with OKT3. Transplantation 1992; 53: 68-72.
Smith SR, Butterly DW, Conlon PJ, Harland RC, Emovon OE. Incidence of cytomegalovirus disease in renal transplantation without antilymphocyte induction: Is prophylaxis necessary? Transplant Proc 1998; 30: 2097-9.
Pouteil-Noble C, Echocard R, Landrivon G and cols. Cytomegalovirus infection: An etiological factor for rejection? A prospective study in 242 cases renal transplant patients. Transplantation 1993; 55: 851-7.
Humar A, Gillingham KJ, Payne WD, Dunn DL, Sutherland DE, Matas AJ. Association between cytomegalovirus disease and chronic rejection in kidney transplant recipients. Transplantation 1999; 68: 1879-83.
Pascual J, Mampaso F, Orofino L and cols. Colorimetric nonisotopic method for in situ hybridization detection of cytomegalovirus in renal allograft biopsies. Transplant Proc 1992; 24 (1): 83-4.
Reploeg MD, Storch GA, Clifford DB. BK virus: a clinical review. Clin Infec Dis 2001; 33: 191-202.
Gardner SD, Mackenzie EFD, Smith C and cols. Prospective study of the human polyomaviruses BK and JC and cytomegalovirus in renal transplant recipients. J Clin Pathol 1984; 37: 578-86.
Boubenider S, Hiesse C, Marchand S and cols. Post-transplantation polyomavirus infections. J Nephrol 1999; 12: 24-9.
Randhawa PS, Finkelstein A, Scantlebury V and cols. Human Polyoma virus-associated intersticial nephritis in the allograft kidney. Transplantation 1999; 67: 103-9.
Drachenberg CB, Beskow CO, Cangro CB and cols. Human polyoma virus in renal allograft biopsies: morphological findings and correlations with urine cytology. Hum Pathol 1999; 12: 24-9.
Rao TSK, Filippone EJ, Nicastri AD and cols. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med 1984; 310: 669-73.
Rao TSK, Friedman EA, Nicastri AD. The types of renal disease in the adquired immunodeficiency syndrome. N Engl J Med 1987; 316: 1062-8.
Bourgoignie JJ. Renal complications of human immunodeficieny virus. Kidney Int 1990; 37: 1571-84.
Cohen AH, Nast CC. HIV-associated nephropathy: a unique combined glomerular, tubular and intersticial lesions. Mod Pathol 1988; 1: 87-97.
Kimmel PL, Phillips TL, Ferreira-Centeno A and cols. HIV-associated immune-mediated renal disease. Kidney Int 1993; 44: 1327-40.
Katz A, Bargman JM, Miller DC and cols. IgA nephritis in HIV-positive patients: a new HIV associated nephropathy? Clin Nephrol 1992; 38: 61-8.
Guerra IL, Abraham AA, Kimmel PL and cols. Nephrotic syndrome associated with chronic persistent hepatitis B in an HIV antibody positive patient. Am J Kidney Dis 1987; 10: 385-8.
Nochy D, Glotz D, Dosquet P and cols. Renal disease associated with HIV infection: a multicentric study of 60 patients from Paris hospitals. Nephrol Dial Transplant 1993; 8: 11-9.
Misiani R, Bellavita P, Fenili D, et al. Hepatitis C virus infection in patients with essential mixed cryoglobulinemia. Ann Intern Med 1992; 117: 573.
Agnello V, Chung RT, Kaplan LM. A role for hepatitis C virus infection in type II cryoglobulinemia. N Eng J Med 1992; 327: 1490.
D’Amico G, Colasanti G, Ferrario F, Sinico RA. Renal involvement in essential mixed cryoglobulinemia. Kidney Int 1989; 35: 1004-14.
Johnson RJ, Gretch DR, Yamabe H and cols. Membranoproliferative glomerulonephritis associated with hepatitis c virus infection. N Engl J Med 1993; 328: 465.
Pouteil-Noble C, Maiza Hakim, Dijourd F, MacGregor B. Glomerular disease associated with hepatitis C virus infection in native kidneys. Nephrol Dial Transplant 2000; 15(suppl): 28.
Stehman-Breen C, Alpers CE, Couser WG, and cols. Hepatitis c vierus and membranous glomerulonephritis. Clin Neprol 1995; 44: 141.
![]()