
Vol. 35, n.º 3, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 3, 2002 |
Luis Fernando Arias, Lucía González, Teresa Álvarez, José Luis Yagüe, Julia Blanco
Departamento de Anatomía Patológica, Hospital Clínico San Carlos, C/Martín Lagos, s/n, 28040 Madrid, España.
La vasculitis puede definirse, de la manera más simple, como una inflamación de la pared vascular. Puede afectar a cualquier órgano. El riñón está comprometido en muchas de ellas; puede ser, en unos casos el órgano más afectado, mientras que en otros, su alteración puede ser leve o aún pasar desapercibida.
El patólogo se encuentra frecuentemente frente a biopsias de pacientes ya con diagnóstico clínico de vasculitis o bien de otros en los que descubre alteraciones histológicas que sugieren un compromiso renal, aún sin tener un cuadro clínico previo que hiciera pensar en ellas. En estas últimas situaciones, el conocimiento de las lesiones microscópicas que puedan presentarse facilitará, la mayoría de las veces, el diagnóstico precoz, que es esencial para el tratamiento correcto y la supervivencia del órgano.
Parece existir un aumento en la frecuencia de las vasculitis en los últimos años, principalmente las microscópicas (1,2).
Aunque las causas de tal incremento no son bien conocidas, podrían estar relacionadas tanto con factores endógenos como exógenos (3,4). Las formas sistémicas han constituido un capítulo complejo de la patología. Tanto su patogenia poco clara como los múltiples criterios utilizados en su diagnóstico se reflejan en las diferentes clasificaciones que se han hecho de ellas.
Aunque no hay una clasificación universalmente aceptada, la más utilizada actualmente es la propuesta en la conferencia para un consenso internacional en Chapel-Hill en 1994 (5). Esta clasificación las divide en tres tipos de acuerdo al tamaño de los vasos predominantemente comprometidos:
Vasculitis de grandes vasos:
— Arteritis (temporal) de células gigantes.
— Arteritis de Takayasu.
Vasculitis de vasos de mediano calibre:
— Poliarteritis nodosa (PAN).
— Enfermedad de Kawasaki.
Vasculitis de pequeños vasos:
— Poliangeítis microscópica.
— Granulomatosis de Wegener (GW).
— Síndrome de Churg-Streuss (SCS).
— Purpura de Henoch-Schönlein.
— Vasculitis crioglobulinémica esencial.
— Angeítis leucocitoclástica cutánea.
Como se ve, el tamaño de vasos afectados es el factor primordial en la nomenclatura. Ahora bien, en la realidad existe interposición de los diferentes tipos de vasculitis. Por ello el diagnóstico requiere la integración de los hallazgos microscópicos con los datos clínicos, inmunológicos y de laboratorio.
Otro abordaje diferente de las vasculitis es el patogénico: infección (por bacterias, hongos o virus); lesión inmunológica (púrpura de Henoch-Schönlein, lupus, vasculitis reumatoidea, Goodpasture, enfermedad de Kawasaki, vasculitis crioglobulinémica, enfermedad del suero, poliarteritis nodosa); pauciinmunes, asociadas o mediadas por anticuerpos citoplasmáticos antineutrófilo: ANCAs (granulomatosis de Wegener, poliangeítis microscópica y síndrome de Churg-Strauss); y las posiblemente mediadas por lesión celular (arteritis de células gigantes, arteritis de Takayasu y lesión vascular del injerto) (4).
Los problemas diagnósticos para el patólogo dimanan del carácter segmentario de la lesión vascular (de ahí la importancia de la cantidad y el tipo de la muestra); del uso de terminología frecuentemente confusa; y de los tratamientos anteriores a la biopsia renal que pueden modificar la imagen histológica y la dificultad de situar cada caso en una de las entidades anatomoclínicas de las clasificaciones usadas. El desconocimiento por parte del patólogo, en muchos casos, de los datos clínicos o del laboratorio del propio paciente, fundamentales para el diagnóstico, incrementan estos problemas.
La etiopatogenia es compleja y no bien conocida en la actualidad. En el caso de la arteritis (temporal) de células gigantes, se postula una reacción inmunológica mediada por células contra un antígeno no conocido en la pared del vaso o contra un antígeno extraño localizado en su pared (6,7). En la arteritis de Takayasu se ha propuesto un mecanismo similar, habiendo encontrado algunos autores relación con el bacilo de la tuberculosis (8,9).
En la enfermedad de Kawasaki se ha sugerido la implicación, aunque no se haya identificado a ninguno de ellos, de un agente infeccioso u otro factor exógeno, por la tendencia a presentarse de una manera endémica o epidémica (10,11). Otros autores han detectado la presencia de anticuerpos que reaccionan con células endoteliales haciendo pensar en una reacción antigeno-anticuerpo in situ (12,13). También en PAN la etiología es desconocida. Se ha propuesto una reacción por complejos inmunes inducidos por infección, detectándose en algunos casos infección por el virus de la hepatitis B (14). En estas dos arteritis de medianos vasos no suelen detectarse ANCAs.
Tampoco es clara la etiología de la poliangeítis microscópica, GW y SCS, aunque se sabe que son enfermedades relacionadas. En ellas se expresan ANCAs que hipotéticamente inducirían inflamación vascular por varios mecanismos: activación directa de neutrófilos en circulación, formación de complejos inmunes in situ o unión a antígenos en las células endoteliales. Aunque más de un mecanismo podría estar implicado, el primero es el más factible dada la ausencia de detección de anticuerpos en las paredes vasculares (4,15-20).
VASCULITIS Y RIÑÓN
Vasculitis de grandes vasos
Excepcionalmente afectan al riñón, pueden comprometer a la arteria renal principal o a la aorta en el ostium renal y ocasionalmente afectan a arterias intraparenquimatosas causando estrechamiento de su luz y produciendo hipertensión renovascular (19,21). Histológicamente la arteritis de células gigantes y la arteritis de Takaysu se caracterizan por inflamación granulomatosa de la pared arterial, más prominente en la capa media, aunque el infiltrado linfohistiocitario se encuentra en todo el espesor del vaso (panarteritis). En la fase activa se identifican células gigantes, granulomas y fragmentación de la lámina elástica; ocasionalmente puede haber necrosis fibrinoide, casi siempre leve y focal, pero en esos casos debe pensarse en otros tipos de vasculitis (4,9,22). El cuadro histológico de las dos arteritis de grandes vasos es muy similar y los hallazgos clínicos son muy importantes en su diferenciación (4). Ha habido algunos informes de lesiones glomerulares en arteritis de Takayasu y de células gigantes, pero tales casos podrían significar dos enfermedades diferentes que coinciden en el mismo paciente, más que tener una relación etiológica directa (21,23-27).
Vasculitis de vasos de mediano calibre
Afectan a las arterias intrarrenales pero no producen, por definición, glomerulonefritis ni vasculitis en arteriolas, capilares ni vénulas (5).
Poliarteritis nodosa
Las lesiones vasculares se localizan en arterias de tamaño grande y mediano, tales como interlobares, arciformes y corticales radiadas (intelobulillares). En la misma biopsia suelen coexistir lesiones en distinto estado evolutivo: agudas y crónicas. Macroscópicamente el riñón puede presentar una superficie de aspecto nodular, que es debido a la existencia de áreas residuales de parénquima hipertrofiado adyacentes a áreas cicatriciales de infarto.
En la fase activa las lesiones histológicas se caracterizan por necrosis fibrinoide segmentaria con infiltración leucocitaria en todo el espesor de la pared (fig. 1b); en fases iniciales los neutrófilos son las células predominantes y frecuentemente se observa leucocitoclasia (fragmentación de neutrófilos en la pared del vaso). La necrosis compromete inicialmente la porción interna de la pared arterial, pero puede hacerse transmural. Característicamente afecta a tramos no continuos, dejando muchos otros respetados, por lo cual pueden requerirse cortes seriados a distintos niveles del tejido para identificar la lesión. Con mayor frecuencia estas lesiones se ubican en las bifurcaciones (4,19). La necrosis fibrinoide tiene el aspecto de fibrina y se ve eosinófila con hematoxilina y eosina y fuschinofílica (roja) con el tricrómico de Masson; se acompaña de picnosis, cariorrexis y ruptura de la lámina elástica (fig. 1c) (28). El endotelio suele estar edematoso o desprendido; pueden encontrarse trombosis en las zonas lesionadas del vaso (fig. 1d) y desarrollarse infartos (fig. 1a, flechas), cuyo tamaño dependerá del calibre del vaso y de la rapidez de la oclusión. En algunos casos se identifican eosinófilos dispersos sin que este hallazgo tenga implicaciones en el diagnóstico (4).
Cuando la necrosis es transmural puede extenderse al tejido perivascular con formación de pseudoaneurismas (fig. 1d). Mucho más rara es la ruptura del vaso con hemorragia perirrenal (fig. 1a) (29,30).
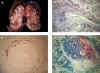
Fig. 1. Panarteritis nodosa.
Al avanzar el proceso los neutrófilos van siendo remplazados por leucocitos mononucleares y la necrosis y leucocitoclasia se hacen paulatinamente menos evidentes. Las lesiones agudas evolucionan hacia la esclerosis con fibrosis de la pared arterial, organización del tejido lesionado y recanalización de trombos, lo que puede llevar a disminución de la luz e hipertensión renovascular (31). El infiltrado inflamatorio adventicial, sin lesión de la pared, no es suficiente para diagnosticar PAN, sino que es necesario hacer cortes seriados para demostrar la lesión en otras capas de la pared (19). La atrofia tubular y fibrosis intersticial son cambios isquémicos asociados.
En un paciente con arteritis necrotizante, el diagnóstico de PAN es adecuado solamente si no hay evidencia de poliangeítis microscópica, GW o SSS. El compromiso de capilares, arteriolas, vénulas o glomérulos (que son capilares) descarta, por definición, el diagnóstico de PAN (14,18,31,32).
Enfermedad de Kawasaki
Es también denominada «síndrome ganglionar mucocutáneo». Habitualmente afecta a niños y se caracteriza por fiebre, linfadenopatía e inflamación de mucosas, de piel y de arterias; de estas últimas, las más afectadas son las coronarias, axilares e ilía cas. Aproximadamente un 25% de los pacientes presenta compromiso de la arteria renal principal. Raras veces se presenta disfunción renal (33). Histológicamente hay edema, infiltración por neutrófilos y mononucleares e inflamación perivascular. AI evolucionar las lesiones va disminuyendo el infiltrado neutrofílico, persistiendo el linfohistiocitario. Hay menos necrosis arterial que en PAN. En las lesiones más evolucionadas aparecen sucesivamente fibroplasia, cicatrización y, en algunos casos, estenosis en el vaso. En el riñón, las arterias interlobares y ocasionalmente las arciformes e interlobulillares están comprometidas, encontrándose necrosis fibrinoide, inicialmente medial (19,33).
VASCULITIS DE PEQUEÑOS VASOS
Son las que más frecuentemente causan disfunción renal, debido principalmente a glomerulonefritis, pero, las lesiones isquémicas también pueden afectar su función. Hay evidencia clínica o patológica de afectación renal en aproximadamente el 90% de pacientes con poliangeítis microscópica, 80% de pacientes con GW y un 45% de pacientes con SCS (20,34,35). Este tipo de vasculitis afecta no solamente vasos de pequeño calibre, sino también, arterias de tamaño mediano. La técnica de inmunofluorescencia permite delimitar dos tipos de vasculitis: las pauciinmunes (Poliangeítis microscópica, Wegener y Churg-Strauss) y las vasculitis con depósitos de inmunoglobulinas (Púrpura de Scholein-Henoch y la vasculitis crioglobulinémica). Así pues, esta técnica es fundamental para diferenciarlas. El estudio serológico para ANCAs es positivo en la mayoría de pacientes con vasculitis pauciinmunes; su demostración también es útil en el diagnóstico diferencial. Estos anticuerpos suelen ser negativos en vasculitis de vasos medianos (19,36,37). En algunos pacientes con glomerulonefritis y positividad para ANCAs, pueden encontrarse también anticuerpos antimembrana basal y para algunos autores no es sólo un epifenómeno, son patogénicos (38-40); de igual manera en 20 a 30% de pacientes con glomerulonefritis proliferativa extracapilar y anticuerpos antiMBG se encuentran ANCAs (15,41).
Características comunes
La lesión histológica común a todas las formas de vasculitis de pequeños vasos es la necrosis fibrinoide segmentaria de la pared arterial, arteriolar, de capilares y de vénulas, es frecuente la infiltración de neutrófilos y leucocitoclasia en diferentes tipos de vasos y a veces se encuentran células gigantes. La inflamación de capilares glomerulares origina glomerulonefritis necrotizante con semilunas, un hallazgo muy característico en estas vasculitis. Las lesiones vasculares son más difíciles de identificar en muchas biopsias, con una frecuencia que varía desde un 11% de casos hasta un 60%, según los diferentes estudios (18,19,32,42,43).
Las semilunas y segmentos necrotizantes glomerulares se identifican en más del 90% de pacientes con glomerulonefritis y ANCAs positivos (43-45). Es común la ruptura de la cápsula de Bowman produciendo un denso infiltrado inflamatorio adyacente, con presencia de células gigantes que, a veces, puede hacer desaparecer el glomérulo y dar el aspecto microscópico de un granuloma en medio del intersticio (fig. 2d). Con tinciones de plata metenamina o Tricrómico de Masson, pueden identificarse fragmentos de membrana basal y aclarar que se trata de restos de un glomérulo (4). Bajema y cols. (46) encuentran que el número de glomérulos normales puede predecir la función renal en pacientes con glomerulonefritis necrotizante asociada a ANCAs.

Fig. 2. Poliangeítis
microscópica.
Las lesiones crónicas se caracterizan por semilunas fibrosas y nefritis intersticial crónica, siendo mayor la fibrosis en las zonas con glomerulos lesionados; en algunos casos habrá intenso infiltrado inflamatorio del intersticio que puede ser una expresión de la lesión de vasos pequeños (fig. 3d). La inflamación de vasos rectos medulares produce nefritis intersticial hemorrágica (19).
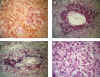
Fig. 3. Poliangeítis
microscópica.
La inmunofluorescencia permite diferenciar las vasculitis pauciinmunes (sin o con pocos depósitos de inmunoglobulinas o complemento) de la glomerulonefritis por anticuerpos anti-membrana basal (IgG lineal en paredes de capilares glomerulares y tubulares) o por inmunocomplejos (depósitos de Ig y complemento).
Poliangeítis microscópica
Es definida por el consenso de Chapel-Hill como una vasculitis necrotizante, sin o con pocos depósitos inmunes, que afecta pequeños vasos y que puede comprometer vasos de mediano calibre y son muy comunes en ella las lesiones glomerulares necrotizantes y la asociación con capilaritis pulmonar (5). Aunque se ha utilizado también el nombre de poliarteritis microscópica, este término no debe ser usado ya que la enfermedad no sólo compromete arterias. El Colegio Americano de Reumatología no reconoce el diagnóstico de poliangeítis microscópica e incluye a tales pacientes en GW, púrpura de Henoch-Schönlein o angeítis por hipersensibilidad (37,47).
Las lesiones necrotizantes vasculares son similares a las de pacientes con GW y SCS y en el riñón la lesión más frecuente es glomerular, caracterizada por necrosis fibrinoide segmentaria (fig. 2c), ruptura de la pared de los capilares (fig. 2b) y formación de semilunas; el número de glomérulos afectados es muy variable; las lesiones se acompañan de un mínimo grado de proliferación endocapilar (fig. 2a) que en ocasiones puede ser muy intenso y las lesiones glomerulares pueden estar en diferentes estados evolutivos en una misma biopsia. Se ven semilunas granulomatosas cuando existe ruptura de la membrana basal de la cápsula de Bowman (fig. 2d). En estos casos, se observan céluas CD-68 positivas en torno al glomérulo (fig. 3a). Los túbulos muestran, al igual que en otras vasculitis de pequeños vasos, frecuentes cilindros y signos de tubulitis (14,18,19,32,38,44,48).
La arteritis en la biopsia renal se observa con poca frecuencia. Jennette sólo la encuentra en un 11% (19). Es necesario realizar cortes seriados del material incluido en parafina. Las arterias afectadas suelen ser a. radiales corticales y arteriolas y con menor frecuencia, a. arcuatas o interlobares. Las lesiones necrotizantes (fig. 3b) o granulomatosas (fig. 3c), son circunferenciales y en el mismo estadio evolutivo. La venulitis o capilaritis en el intersticio produce un intenso infiltrado inflamatorio constituido por linfocitos, células plasmáticas, neutrófilos y a veces, eosinófilos, así como focos de hemorragia (fig. 3d).
Los vasos extrarrenales más frecuentemente afectados son arterias y arteriolas de intestino, músculo esquelético, corazón y bazo, vénulas y arteriolas de piel y capilares pulmonares (19). Algunos pacientes presentan una lesión glomerular idéntica, sin vasculitis extraglomerular, que ha sido considerada una variante, limitada al riñón, de poliangeítis microscópica (18,49)
La gran mayoría de pacientes tienen ANCAs positivos y pueden presentar anticuerpos antimieloperoxidasa (MPO) o antiproteinasa 3 (PR3), aunque se ha dicho que es más frecuente la expresión de P-ANCAs (anti-MPO) (14,18,20,37,42).
Granulomatosis de Wegener
Es definida por el consenso de Chapel-Hill como una inflamación granulomatosa que compromete el tracto respiratorio, con vasculitis necrotizante que afecta a vasos de pequeño y mediano calibre y que frecuentemente presenta glomerulonefritis necrotizante (5). Este criterio restrictivo excluye de esta categoría a aquellos casos que tienen inflamación no granulomatosa del aparato respiratorio. Probablemente el 80% de pacientes con GW desarrollarán compromiso renal en algún momento de su evolución (16).
La lesión más característica en el tracto respiratorio es inflamación granulomatosa, necrotizante, focal, irregular, con neutrófilos dispersos; la capilaritis alveolar es idéntica a la encontrada en poliangeitis microscópica, excepto, que es acompañada por inflamación granulomatosa (18,19).
Fuera del tracto respiratorio es más común la vasculitis que los granulomas. Así, en el riñón será menos frecuente encontrar lesión granulomatosa; allí, la glomerulonefritis y vasculitis son también histológicamente idénticas a las de poliangeítis microscópica (figs. 4a y 4b). Cuando hay ruptura de la membrana basal capsular, se observa una inflamación con macrófagos epitelioides, linfocitos y células gigantes, formando un granuloma periglomerular relativamente esférico (figs. 4c y 4d). Estos granulomas pueden verse en otras glomerulonefritis extracapilares, con ruptura capsular, como las producidas por anticuerpos anti-membrana basal o por inmunocomplejos, aunque son más frecuentes en las pauciinmunes y más aún en la GW, por lo cual no es específico de esta última (19,50). Los granulomas necrotizantes intersticiales (fig. 5c), no asociados a glomérulos, son mucho más inusuales y son un componente de la granulomatosis sistémica y sí indican GW. A bajo aumento son más irregulares y menos bien definidos (4). La técnica de inmunofluorescencia demuestra la presencia exclusiva en la mayor parte de los casos, de fibrinógeno (fig. 5a).

Fig. 4. Granulomatosis de Wegener.
Las lesiones arteriales, son similares a las de la poliangeítis microscópica, necrotizantes o granulomatosas, con presencia de células gigantes multinucleadas (fig. 5b).
El criterio más fiable para diferenciar esta vasculitis de la poliangeítis microscópica es una cuidadosa correlación clinicopatológica, con la demostración de inflamación granulomatosa del aparato respiratorio. Los granulomas en esta localización son poco compactos, con leucocitos dispuestos en empalizada y células gigantes dispersas, diferentes de los granulomas compactos de la tuberculosis o de la sarcoidosis (fig. 5d) (4). La dificultad de demostrar este tipo de inflamación nos crea uno de los problemas diagnósticos más importantes; en algunos casos su búsqueda muestra resultados negativos y en otros casos es necesario que transcurra un tiempo hasta que se compruebe la participación inflamatoria del aparato respiratorio y su carácter granulomatoso. En aquellos casos en que no se demuestre este tipo de patrón inflamatorio, se recomienda considerarlos inicialmente como poliangeítis microscópica aunque este diagnóstico tenga que modificarse posteriormente (5).
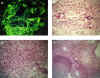
Fig. 5. Granulomatosis de Wegener.
Se encuentran ANCAs en alrededor del 90% de casos, la mayor parte (80%) tiene anticuerpos anti-PR3 y 20% anti-MPO (37,42,43).
Síndrome de Churg-Strauss
Es definido como una inflamación granulomatosa rica en eosinófilos, que compromete el tracto respiratorio y que presenta vasculitis necrotizante de pequeños y medianos vasos y está asociado a asma y eosinofilia (5).
Las lesiones vasculares y glomerulares pueden ser idénticas a las de la poliangeítis microscópica y GW, aunque, típicamente hay un infiltrado de eosinófilos más prominente (fig. 6b),es un hallazgo característico pero no exclusivo de SCS, ya que también pueden encontrarse eosinófilos en otros tipos de vasculitis (19).
La mayoría de los pacientes presenta enfermedad renal leve, siendo raro el compromiso severo. Las lesiones glomerulares suelen ser segmentarias y con semilunas (figs. 6a y 6c), pero, generalmente en pocos glomérulos. Las lesiones vasculares suelen ser también necrotizantes y/o granulomatosas, siendo llamativo el predominio de eosinófilos (4,19).
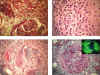
Fig. 6. A, B y C: Granulomatosis de
Churg-Strauss. D: Glomerulonefritis paucinmune.
Los ANCAs son positivos en cerca de un 75% de casos, predominando los anti-MPO (P-ANCAS) (37,51,52).
Glomerulonefritis paucinmune (Glomerulonefritis proliferativa epitelial tipo lIl)
Es una opinión cada vez mas compartida por diferentes autores, que este tipo de Glomerulonefritis, representa una forma de vasculitis limitada al riñon y localizada en los capilares glomerulares fundamentalmente. La frecuente presencia de ANCAs, predominando los anti-MPO, apoya esta teoría.
Vasculitis mediadas por complejos inmunes
El tipo de vasculitis tiene una expresión morfológica muy variable. La glomerolonefritis que se produce usualmente muestra características proliferativas o membranoproliferativas, más que el tipo de lesiones necrotizantes de las vasculitis pauciinmunes (38). Sin embargo, las lesiones glomerulares pueden ser indistinguibles por microscopia de luz.
En la púrpura de Henoch-Schönlein la vasculitis necrotizante es poco habitual y las lesiones glomerulares suelen mostrar proliferación endocapilar, menos frecuentemente semilunas y se detecta IgA en el mesanglo (4,38). Los cambios glomerulares en la nefritis lúpica presentan un amplio espectro, desde ninguna alteración por microscopia de luz hasta proliferación difusa, engrosamiento de membranas basales o esclerosis global. Las lesiones glomerulares se acompañan de vasculitis necrotizante en muchos casos. En las vasculitis crioglobulinémicas la expresión glomerular más común es una glomerulonefritis membranoproliferativa tipo I y la inmunofluorescencia demostrará depósitos de diferentes Igs y complemento (53,55).
CONCLUSIONES
Cualquier vasculitis sistémica puede afectar el riñón. Las de pequeños vasos son las que más frecuentemente lo hacen, siendo la lesión renal más prominente la glomerulonefritis, que en muchos de los casos se acompaña de proliferación epitelial y segmentos necrotizantes. La causa más frecuente del síndrome riñón-pulmón son las vasculitis pauciinmunes. El diagnóstico adecuado de las lesiones renales debe estar basado en una correlación de los hallazgos histológicos y de inmunofluorescencia, con la información clínica y serológica.
BIBLIOGRAFÍA
Andrews M, Edmunds M, Campbell A, Walis J, Feehally J. Systemic vasculitis in the 1980s: Is there an increasing incidence of Wegener’s granulomatosis and microscopic polyarteritis? J R Coll Phys Lond 1990; 24: 284-8.
Haes M, Spargo BH, Wit EJ, Meehan SM. Etiologies and outcome of acute renal insufficiency in older adults. A renal biopsy study of 259 cases. Am J Kidney Dis 2000; 35: 433-47.
Dolman KM, Gans RO, Vervaat TJ y cols. Vasculitis and antineutophil cytoplasmic autoantibodies associated with propylthiouracil therapy. Lancet 1993; 342: 651-2.
Jennette JC, Olson JL, Schwartz MM, Silva FG. (Eds.). Heptinstall’s Pathology of the Kidney. 5.ª ed. Lippincott-Raven, Philadelphia, 1998.
Jennette JC, Falk RJ, Andrassy K y cols. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Reum 1994; 37: 187-92.
Cid MC, Campo E, Ercilla G y cols. Immunohistochemical analysis of Iymphoid and macrophege cell subsets and their immunologic activation markers in temporal arteritis: influence of corticosteroid treatment. Arthritis Rheum 1989; 32: 884-93.
Shiiki H, Shimokama T, Watanabe T. Temporal arteritis: cell composition and the possible pathogenetic role of cell-mediated immunity. Hum Pathol 1989; 20: 1057-64.
Aggarwal A, Cheg M, Sinha M, Nalk S. Takayasu’s arteritis: role of mycobacterium tuberculosis and its 65 kDa heat shock protein. Int J Cardiol 1996; 55: 49-55.
Sharma BK, Jain S, Radotra BD. An autopsy study of Takayasu arteritis in India. Int J Cardiol 1998; 66(supl): 85-90.
Bell DM, Brink EW, Nitzkin JL y cols. Kawesaki syndrome: description of two outbreaks in the United States. N Engl J Med 1981; 304: 1568-1575.
Burns JC, Kushner Hl, Bastian JF y cols. Kawasaki disease. A brief history. Pediatrics 2000; 106: E27.
Leung DY. Immunologic aspects of Kawasaki syndrome. J Rheumatol 1990 (supl); 24: 15-8.
Leung DY, Collins T, Lapierre LA, Geha RS, Pober JS. Immunoglobulin M antibodies present in the acute phase of Kawasaki syndrome Iyse cultured vascular endothelial celis stimulated by gamma interferon. J Clin Invest 1986; 77: 1428-35.
Gayraud M, Gulilevin L, Le Toumelin P y cols. Long-term follow-up of polyarteritis nodosa, microscopic polyangiitis, and Churg-Streuss syndrome: analysis of four prospective triais including 278 patients. Arthritis Rheum 2001; 44: 666-75.
Bosch X, Mirapeix E, Font J y cols. Prognostic implication of anti-neutrophil cytoplasmic autoantibodies with myeloperoxidase specificity in antiglomerular basement membrane disease. Clin Nephrol 1991; 36: 107-13.
Duna GF, Galperin C, Hoffman GS. Wegener’s granulomatosis. Rheum Dis Clin North Am 1995; 21: 949-86.
Harper L, Cockwell P, Adu D, Savage CO. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int 2001; 59: 1729-38.
Jennette JC, Thomas OB, Falk RJ. Microscopic polyangiitis (microscopic polyarteritis). Semin Diagn Pathol 2001; 18: 3-13.
Jennette JC, Falk RJ. The pathology of vasculitis involving the kidney. Am J Kidney Dis 1994; 24: 130-41.
Lhote F, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: clinical aspects and treatment. Rheum Dis North Am 1995; 21: 911-47.
Lenz T, Schmidt R, Scherberich JE, Grone HJ. Renal failure in giant cell vasculitis. Am J kidney Dis 1998; 31: 1044-7.
Numano F, Okawara M, Inomata H, Kobayeshi Y. Takayasu’s arteritis. Lancet 2000; 356: 1023-5.
Cavatorta F, Campisi S, Trabassi E, Zollo, A, Salvidio G. IgA nephropathy associated with Takayesu’s arteritis: report of a case and review of the literature. Am J Nephrol 1995; 15: 165-7.
Lai KN, Chan KW, Ho CP. Glomerulonephritis associated with Takayasu’s arteritis: report of three cases and review of literature. Am J Kidney Dis 1986; 7: 197-204.
Logar D, Rozman B, Vizjak A, Ferloga D, Mulder AHX, Kallenberg CG. Arteritis of both carotid arteries in a patient with focal, crescentic glomerulonephritis and anti-neutrophil cytoplasmic autoantibodies. Br J Rheumatol 1994; 33: 167-9.
Sonnenblick M, Nesher G, Rosin A. Nonclassical organ involvement in temporal arteritis. Semin Arthritis Rheum 1989; 19: 183-90.
Yoshimura M, Kida H, Salto Y y cols. Peculiar glomerular lesions in Takayasu’s arteritis. Clin Nephrol 1985; 24:120-7.
Bajema IM, Bruijn JA. What stuff is this? A historical perspective on fibrinoid necrosis. J Pathol 2000; 191: 235-38.
Maes K, Billiet I, Haerens M, Mattelaer J. Massive bilateral renal and perirenal hemorrhege due to polyarteritis nodosa: a life-threatening uroligic condition. Eur Urol 2000; 38: 349-51.
Zapzalka DM, Thompson HA, Borowsky SS, Coleman-Steenson CC, Mahowald ML, O’Connell KJ. Polyerteritis nodosa presenting as spontaneous bilateral perinephric hemorrhage. Management with selective arterial embolization. J Urol 2000; 164: 1294-5.
Bakkaloglu SA, Ekim M, Tumer N, Tulunay O, Ozer T. Severe renal impairment in the case of classic polyerteritis nodosa. Pediatr Nephrol 2001; 16: 148-50.
Kirkland GS, Savige J, Wilson D, Heale W, Sinclair RA, Hope RN. Classical polyarteritis nodosa and microscopic polyarteritis with medium vessel involvement. A comparison of the clinical and laboratory features. Clin Nephrol 1997; 47: 176-80.
Foster BJ, Bernard C, Drummond KN. Kawasaki disease complicated by renal artery stenosis. Arch Dis Child 2000; 83: 253-5.
Hoffman HS, Kerr GS, Leavitt RY y cols. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116: 488-98.
Nachmant PH, Hogan SL, Jennett JC, Falk RJ. Treatment response and relapse in ANCAassociated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7: 33-9.
Falk RJ, Nachman PH, Hogan SL, Jennette JC. ANCA glomerulonephritis and vasculitis: a Chapel-Hill perspective. Semin Nephrol 2000; 20: 233-43.
Savige J, Davies D, Falk RJ, Jennette JC, Wiik A. Antineutrophil cytoplasmic antibodies and associated diseases: A review of the clinical and laboratory features. Kidney Int 2000; 57: 846-62.
Harris AA, Falk RJ, Jennette JC. Crescentic glomerulonephritis with a paucity of glomerular immunoglobulin localization. Am J Kidney Dis 1998; 32: 1179-84.
Short AK, Esnault VLM, Lockwood M. Anti-neutrophil cytopiasm antobodies and antiglomerular basement membrane antibodies: two coexisting distinct autoreactivities detectable in patients with rapidly progessive glomerulonephritis. Am J Kidney Dis 1995; 26: 439-45.
Tang S, Chan KW, Chan TM, Lui SL, Cheng IK. Anti-glomerular basement membrane and antineutrophil cytoplasm antobody-positive vasculitis presenting with peripherai neuropathy and acute renal failure. Nephron 1998; 79: 225-6.
Verburgh CA, Bruijn JA, Daha MR, Van Es LA. Sequential development of anti-GBM nephritis and ANCA-associated pauci-immune glomeruloneprhitis. Am J Kidney Dis 1999; 34: 344-8.
Jennette JC, Falk RJ. Antineutrophil cytoplasmic autoantibodies and associated diseases: a review. Am J Kidney Dis 1990; 15: 517-29
Jennette JC, Wilkman AS, Falk RJ. Anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and vasculitis. Am J Pathol 1989; 135: 921-30.
D’Agati V, Chander P, Nash M, Mancilla-Jimenez R. Idiopathic microscopic polyarteritis nodosa: ultrastructural observations on the renal vascular and glomerular lesions. Am J Kidney Dis 1986; 7: 95-110.
Inoue M, Akikusa B, Masuda Y, Kondo Y. Demostration of microaneurysms at the interlobular arteries of the kidneys in microscopic polyangiitis: a threedimensional study. Hum Pathol 1998; 29: 223-7.
Bajema lM, Hagen EC, Hermans J y cols. Kidney biopsy as a predictor for renal outcome in ANCA-associated necrotizing glomerulonephritis. Kidney Int 1999; 56: 1751-8.
Leavitt RY, Fauci AS, Bloch DA y cols. The American College of Rheumatology 1990 criteria for the classification of Wegener s granulomatosis. Arthritis Rheum 1990; 33: 1101-7.
Hauer HA, Bajema IM, de Heer E, Hermans J, Hagen EC, Bruijn JA. Distribution of renal lesions in idiopathic vasculitis: a three-dimensional analysis of 87 glomeruli. Am J Kidney Dis 2000; 36: 257-65.
Croker BP, Lee T, Gunnelis JC. Clinical and pathologic features of polyarteritis nodosa and its renal-limited variant: primary crescentic and necrotizing glomerulonephritis. Hum Pathol 1987; 18: 38-44.
Bhathena DB, Migdel SD, Julian BA, McMorrow RG, Baehler RW. Morphologic and immunohistochemical observations in granulomatous glomerulonephritis. Am J Pathol 1987; 126: 581-91.
Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999; 78: 26-37.
Kikuchi Y, Ikehata N, Tajima O, Yoshizawa N, Miura S. Glomerular lesions in patients with Churg-Strauss syndrome and the antimieloperoxidase antibody. Clin Nephrol 2001; 55: 429-35.
D’amico G. Renal involvement in hepatitis C infection: cryoglobulinemic glomerulonephritis. Kidney Int 1998; 54: 650-71.
Lamprecht P, Gause A, Gross WL. Cryoglobulinemic vasculitis. Arthritis Rheum 1999; 42: 2507-16.
![]()