
Vol. 35, n.º 3, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 3, 2002 |
María Luz Picazo García, Belén Pérez-Mies, Isabel Esteban Rodríguez
Departamento de Anatomía Patológica. Hospital Universitario «La Paz». Madrid.
En 1925 Moschowitz (1) describe el caso de una niña de 16 años que debuta con un cuadro clínico agudo con fiebre, anemia, púrpura y manifestaciones neurológicas. En la autopsia se demostraron trombos hialinos en las arteriolas y en los capilares del corazón, higado y riñón. En 1947 Singer (2) propone para este cuadro clínico el término de Púrpura Trombótica Trombocitopénica (PTT).
En 1952 Symmers (3) utiliza por primera vez el término de Microangiopatía Trombótica (MAT) para describir las lesiones vasculares que acompañan a la PTT.
En 1955 Gasser (4) utiliza el término de Síndrome Hemolítico Urémico (SHU), para describir un cuadro clínico agudo en niños, caracterizado por anemia hemolítica, trombocitopenia e insuficiencia renal con necrosis cortical difusa de los riñones. Con posterioridad, Habib (5) describe los cambios histológicos glomerulares y arteriolares que acompañan al SHU infantil y propone también el término de MAT para definir a estas lesiones.
En ambos casos, el término MAT se utiliza para definir un proceso patológico caracterizado por el engrosamiento de las paredes de las arterias pequeñas, arteriolas y capilares y por la presencia de trombos intraluminares de plaquetas y fibrina. Las paredes vasculares presentan tumefacción y descamación del endotelio y acúmulo de un material mucoide, translucido, en el espacio subendotelial. Estas lesiones producen una oclusión parcial o total de la luz de los pequeños vasos.
A pesar de que las lesiones morfológicas de la PTT y del SHU son similares, de tipo microangiopático, clásicamente los dos síndromes han sido considerados como entidades diferentes debido a que presentan distintas manifestaciones clínicas.
Así, la PTT idiopática clásica, es una enfermedad esporádica, que afecta a los adultos, con predominio de las mujeres (relación mujer-hombre de 3:2) y su mayor incidencia está entre la 3.ª y la 4.ª década de la vida. La PTT es una enfermedad aguda que con frecuencia se acompaña de un síndrome constitucional inespecífico, con malestar, nauseas y vómitos. Aproximadamente el 75% de los pacientes presentan anemia hemolítica microangiopática, trombocitopenia y síntomas neurológicos y es prácticamente constante la fiebre. La afectación renal puede ser ligera (6-8).
Por el contrario, el SHU clásicamente se ha descrito como un síndrome agudo, que afecta a los niños en los primeros años de la vida, sin preferencias de sexos. El cuadro sindrómico clásico de anemia hemolítica, trombocitopenia e insuficiencia renal aguda, suele estar precedido por síntomas gastrointestinales con vómitos, diarrea y dolor abdominal. En más del 90% de los niños existe insuficiencia renal aguda y hasta en un 40% de los casos existen síntomas y signos neurológicos.
Desde la descripción inicial de estos dos síndromes hasta la actualidad, numerosos trabajos han confirmado que la PTT no es una enfermedad limitada a los adultos (8) ni el SHU afecta sólo a los niños como inicialmente se pensaba. Es cierto, que la forma epidémica clásica de SHU, es más frecuente en la infancia, en los primeros años de la vida, pero también se ha descrito en los adultos (9,10).
Por otra parte, la afectación neurológica no es exclusiva de la PTT ya que se sabe que es una de las causas más frecuentes de mortalidad en la fase aguda del SHU infantil (11). Además, la afectación renal no sirve para diferenciar el SHU de la PTT, ya que más del 80% de los pacientes con PTT tienen afectación renal y entre el 40% y el 80% presentan disminución de la función renal (7,8).
Por todos estos datos, algunos investigadores consideran a la PTT y al SHU como una misma enfermedad, con idéntica lesión patológica de base, pero con diferente expresión clínica y han propuesto utilizar el término SHU/PTT para describir el cuadro clínico que acompaña a los pacientes con MAT (12,13).
HALLAZGOS DE LABORATORIO
Las manifestaciones clínicas que acompañan a la MAT pueden ser variables y diversas dependiendo del órgano más afectado y del número de vasos lesionados, pero siempre los pacientes presentan trastornos hematológicos con anemia hemolítica y trombocitopenia La anemia hemolítica microangiopática, es el dato analítico esencial para el diagnóstico del SHU/PTT y es un hallazgo constante en todas las situaciones clínicas que cursan con lesiones vasculares de tipo microangiopático. Este tipo de anemia se caracteriza por la presencia en la circulación de hematíes deformados y fragmentados (esquistocitos) con un test de Coombs negativo. Los hematíes se fragmentan y se destruyen mecánicamente al atravesar los pequeños vasos lesionados. La trombocitopenia es un hallazgo constante y puede ser severa y es consecuencia de la destrucción y del consumo de las plaquetas en los vasos lesionados. Además, existe elevación de lacticodeshidrogenasa (LDH) y de bilirrubina indirecta y disminución de los niveles séricos de haptoglobina, todos ellos marcadores de hemólisis intravascular.
ETIOPATOGENIA
La etiopatogenia de la lesión vascular del SHU/PTT es compleja, pero la lesión del endotelio es el factor básico que precede y mantiene el desarrollo de las lesiones microangiopáticas. En la actualidad, se conoce que son muchos los agentes que han sido implicados en el desarrollo de las lesiones de la microcirculación , todos ellos potencialmente tóxicos para el endotelio vascular (6,7,13,14).
AGENTES INFECCIOSOS: Desde la descripción en 1955 del SHU (4) hasta la actualidad, numerosos estudios epidemiológicos y clínicos han demostrado que existe una estrecha relación entre los brotes epidémicos de enterocolitis causada por E. coli y el desarrollo del SHU en los niños pequeños. Las investigaciones realizadas en las dos últimas décadas sobre esta asociación, han demostrado que determinadas toxinas bacterianas, están directamente relacionadas con el desarrollo del SHU epidémico. Se ha confirmado que determinadas cepas de E. Coli (por ejemplo el tipo O157:H7) producen una toxina similar a la toxina Shiga producida por la Shigella dysenteriae tipo I. Esta toxina, denominada «toxina tipo Shiga» o Verocitotoxina (porque en los cultivos lesionan a las células Vero), tiene un efecto citotóxico directo sobre los endotelios, sobre todo en presencia del Factor de Necrosis Tumoral (TNF) y de la interleukina IL-1 (7,13,14).
El SHU/PTT esporádico que aparece en niños mayores y en adultos, se asocia con menor frecuencia a infecciones bacterianas productoras de citotoxinas (S. dysenteriae, Salmonela typhi, Strectococo pneumoniae etc.) (13).
En ocasiones, la MAT se ha desrito asociada con infecciones víricas (15) y con el síndrome de inmunodeficiencia adquirida (HIV) (16,17).
COMPLEJOS INMUNES: En el suero de algunos pacientes con SHU/PTT, se han encontrado anticuerpos que son citotóxicos para las células endoteliales, lo que sugiere que algunos complejos inmunes pueden tener un papel patogénico en el desarrollo de la MAT (13). Esto explicaría también, la presencia de lesiones microangiopáticas en enfermedades con base inmunológica. Así, la MAT se ha descrito asociada al LES (18-20), en el Síndrome antifosfolípido primario (21), en las Glomerulonefritis (GN) asociadas con ANCA (22) y en GN por anticuerpos anti-MB glomerular (23).
FÁRMACOS: Numerosos agentes quimioterápicos (7,14,24,25) e inmunosupresores (Ci closporina A y Tacrolimus) (26-30) han sido implicados en el desarrollo del SHU/PTT. El SHU/PTT se desarrolla en el 5%-10% de los pacientes tratados con mitomycina C y las manifestaciones clínicas están relacionadas con la dosis acumulativa del medicamento.
En la preeclampsia y en el fracaso renal postparto, se han descrito alteraciones morfológicas, clínicas y de laboratorio similares a las descritas en el SHU/PTT, por lo que muchos investigadores consideran a estas entidades, como una forma de MAT. En algunos casos, ciertas infecciones, algunos medicamentos utilizados durante el parto y la asociación con enfermedades inmunológicas, han sido implicadas en el desarrollo del SHU/PTT postparto (7).
La existencia de casos familiares de SHU/PTT, en los que los episodios recurrentes de la enfermedad son mucho más frecuentes que en los casos no familiares, sugiere que alteraciones genéticas, que se acompañan con niveles séricos disminuidos de C3 y con déficit de Factor H pueden predisponer al desarrollo de la MAT (31,32).
En el momento actual, de todos los posibles agentes etiológicos implicados en el desarrollo de la MAT causal, sólo se ha podido demostrar una relación etiológica entre la Verocitotoxina y el SHU epidémico asociado con diarrea (7,13,14).
La lesión del endotelio, desencadenada por cualquier agente etiológico, origina alteraciones en sus propiedades anticoagulantes, disminuye su actividad fibrinolítica y facilita la exposición de la superficie subendotelial, que es trombogénica.
En condiciones normales, las células endoteliales sintetizan muchas sustancias que intervienen en la coagulación y en la fibrinolisis, tales como la prostaciclina (PGI2), (potente inhibidor de la agregación plaquetaria), Óxido Nítrico (NO) (vasodilatador) factor de von Willebrand (agregante plaquetario) y tromboxano A2. La lesión del endotelio, disminuye la producción de prostaciclina , desestabiliza el balance entre la PGI2 y tromboxano, disminuye la síntesis de NO y favorece la liberación a la sangre de multímeros excesivamente grandes de factor de v. Willebrand. Todos estos factores, favorecen la agregación plaquetaria, la formación de trombos intraluminares y producen vasoconstricción con la consiguiente lesión vascular y daño orgánico. Las plaquetas agregadas y activadas en las luces de los pequeños vasos, liberan factores de crecimiento (FGDP), que estimulan la proliferación fibroblástica y producen engrosamiento fibroso de la íntima vascular (6,13).
HALLAZGOS PATOLÓGICOS
En la MAT, el aspecto macroscópico de los riñones es variable en función de la intensidad y de la duración de la enfermedad. En la fase aguda, el riñón puede estar aumentado de tamaño, edematoso, congestivo, con petequias y pequeñas hemorragias corticales. Además, en los casos más graves, pueden verse infartos parcheados o una extensa necrosis cortical.
En las etapas más evolucionadas, el riñón puede ser normal o estar disminuido de tamaño, con cicatrices parcheadas y con depósitos de calcio en relación con las zonas previamente necrosadas.
De acuerdo con las descripciones clásicas de Habib y col. (5), las lesiones histológicas renales pueden ser variables y afectar preferentemente a los glomérulos (MAT glomerular), afectar a las pequeñas arterias (MAT vascular) o expresarse como una necrosis cortical.
En la MAT glomerular, los glomérulos están siempre afectados, pero en los casos más benignos, la afectación puede ser transitoria. Además, la distribución de las lesiones es variable de unos glomérulos a otros, dependiendo de la gravedad y de la duración de la enfermedad. La lesión glomerular típica descrita por Habib (5,33,34) se observa fundamentalmente en niños por debajo de los 2 años de edad, con SHU clásico asociado con diarrea. En las etapas iniciales, los glomérulos presentan un engrosamiento de la pared capilar, con tumefacción y descamación del endotelio. Hay edema del espacio subendotelial y acumulación de un material fibrino-mucoide traslucido. A veces, existe neoformación de membrana basal e interposición de células mesangiales lo que origina imágenes de dobles contornos . En la luz de los capilares glomerulares pueden verse hematíes fragmentados, fibrina y trombos de plaquetas. La matriz mesangial está edematosa con aspecto finamente reticular, pero sin incrementos significativos de la celularidad mesangial (figs. 1 y 2).
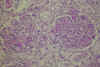
Fig. 1. MAT
glomerular: tumefacción endotelial, edema subendotelial, trombos intracapilares
y aspecto fibrilar de la matriz mesangial (PAS).
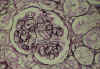
Fig. 2. MAT
glomerular, las asas capilares están engrosadas con dobles contornos
traslúcidos. El área mesangial no está engrosada.
En los casos más graves, puede verse desaparición parcial o total de las células y de la matriz mesangial, con capilares glomerulares elongados y dilatados, vacíos u ocupados por proteínas, hematíes o trombos. Estas dilataciones capilares están producidas por la separación de las membranas basales, de la matriz mesangial degenerada (mesangiolisis) (fig. 3).

Fig. 3. MAT
glomerular, las luces capilares están ectásicas, vacías o con trombos de
fibrina (mesangiolisis) (HE).
Los trombos intraluminares de plaquetas y fibrina, se ven sólo en algunos glomérulos y suelen estar ausentes en las formas clínicas menos graves. En general, la trombosis comienza en el hilio y se extiende a las zonas periféricas del ovillo capilar, pero rara vez afecta a la totalidad del glomérulo. Si la lesión glomerular es importante, se pueden producir áreas focales o extensas de necrosis cortical y depósitos cálcicos.
En ocasiones, pueden verse pequeñas semilunas, infiltrados de leucocitos polimorfonucleares y ligera proliferación mesangial, lo que origina una imagen semejante a la GN proliferativa difusa endocapilar.
Las lesiones agudas leves de las paredes capilares y del mesangio, pueden regresar a medida que la enfermedad se resuelve, pero las lesiones más intensas, evolucionan hacia una forma peculiar de esclerosis glomerular «pálida», caracterizada por su apariencia fibrilar y por conservar en parte la arquitectura lobular del glomérulo (fig. 4).

Fig. 4. MAT
glomerular, lesión crónica. Aspecto fibrilar del ovillo capilar, con
disminución del número de células (T. de Masson).
En las etapas más avanzadas de la enfermedad, las alteraciones glomerulares son de tipo crónico. Puede verse ampliación del mesangio con esclerosis mesangial, engrosamientos focales de las paredes de los capilares glomerulares con interposición de células mesangiales e imágenes de dobles contornos, lo que origina una lesión glomerular similar a la de una GN membranoproliferativa.
La MAT arterial afecta a las arterias interlobulares y a las arteriolas. En estadios iniciales, la luz arteriolar está ocluida por expansión mucoide del espacio subendotelial (fig. 5). Además, pueden verse depósitos de fibrina limitados a la íntima o con extensión a la pared vascular (fig. 6), acompañándose en ocasiones, de infiltrados de polimorfonucleares. Pueden verse trombos intraluminares, hematíes fragmentados, proliferación miointimal y a veces, necrosis fibrinoide de la pared arteriolar. A veces, las arteriolas presentan dilataciones aneurismáticas o forman estructuras glomeruloides secundarias a la proliferación endotelial (fig. 7).
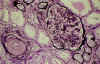
Fig. 5. MAT
arterial, intenso edema mucoide subendotelial, con oclusión de la luz
arteriolar. Algunas asas capilares del glomérulo presentan plegamiento
isquémico (Metenamina plata).

Fig. 6. MAT
arterial, arteriolas con edema subendotelial y trombos de fibrina intraluminares
y subendoteliales (T. de Masson).
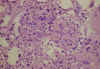
Fig. 7. Estructura
"glomeruloide", secundaria a la proliferación endotelial
intraluminal, en las arteriolas del hilio glomerular (HE).
Las arterias interlobulares, presentan tumefacción endotelial, asociada a veces, a la presencia de fibrina en el espacio subendotelial, en la pared vascular. Existe edema mucoide subendotelial y pueden verse hematíes fragmentados en la luz vascular y en la capa media muscular lo que ocasiona una marcada reducción de la luz vascular. Con el paso del tiempo, estas lesiones agudas evolucionan a la cronicidad. La íntima se engruesa por proliferación de células fibromioides, que se disponen en capas concéntricas «hojas de cebolla». Estas alteraciones vasculares originan cambios isquémicos glomerulares, caracterizados por retracción del ovillo capilar, engrosamiento y plegamiento de las paredes de los capilares y dilatación microquística del espacio urinario (fig. 8). En ocasiones, las alteraciones isquémicas afectan difusamente a la totalidad de los glomérulos, originándose una imagen histológica de Enfermedad Renal Glomeruloquística (observación personal, 35).
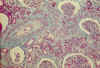
Fig. 8. MAT
arterial, proliferación fibromioide intimal de la arteria interlobular. En la
arteriola, la proliferación se dispone en capas concéntricas. Los ovillos
glomerulares presentan cambios isquémicos con ampliación del espacio urinario
(T. Masson).
Los capilares glomerulares, pueden presentar dilatación y congestión de las luces capilares (parálisis glomerular), áreas segmentarías de necrosis, hematíes fragmentados intracapilares, proliferación extracapilar focal y en los casos más intensos, asociarse con lesiones típicas de MAT glomerular.
En la MAT, los túbulos presentan cilindros hialinos y cilindros hemáticos. En los casos más graves, el epitelio tubular presenta cambios degenerativos y áreas focales de necrosis. En el intersticio hay edema, infiltrados inflamatorios de linfocitos y fibrosis.
Con inmunofluorescencia, en los glomerulos existen sistemáticamente depósitos de fibrinógeno a lo largo de la pared de los capilares glomerulares y en los ejes mesangiales (fig. 9). Se pueden ver también depósitos granulares de IgM, C3 y C1q en las paredes de los capilares, siendo más infrecuentes los depósitos de IgA e IgG. En las paredes de las arterias pequeñas y arteriolas, hay depósitos de fibrina , IgM, C3 y a veces C1q. Los trombos intravasculares muestran depósitos con fibrinógeno y con fibrina.

Fig. 9. MAT
glomerular. Con antisuero antifibrinógeno se observan depósitos en las paredes
de los capilares glomerulares y en los trombos capilares.
En el estudio ultraestructural, los capilares glomerulares presentan células endoteliales hinchadas con citoplasmas pálidos, con escasas organelas. Algunos capilares pueden tener denudación del endotelio con los hematíes, leucocitos y un material electrodenso contactando directamente con la membrana basal glomerular. El dato más característico es el engrosamiento de la pared de los capilares por expansión del espacio subendotelial, que está ocupado por un material pálido finamente granular, correspondiente a fibrina y restos celulares, rodeados de una fina capa de material tipo membrana basal.
En las descripciones clásicas de la PTT se recoge que las alteraciones glomerulares son leves. Algunos glomérulos presentan microtrombos y a veces las paredes de los capilares muestran engrosamientos focales. Sin embargo, son constantes los trombos granulares, eosinófilos, en las luces de las arterias interlobulares y en las arteriolas. Con frecuencia, los trombos ocluyen totalmente la luz vascular y se extienden y se incorporan a las paredes vasculares. En la PTT clásica, son además frecuentes las estructuras glomeruloides en el hilio glomerular.
En la MAT, además de las lesiones renales, se pueden ver trombos intraluminares y lesiones arteriales y arteriolares en múltiples órganos tales como cerebro, tracto gastrointestinal, piel, músculo esquelético, páncreas, corazón, bazo y glándulas suprarrenales.
El estudio de biopsias seriadas ha demostrado que las lesiones bien desarrolladas de la MAT glomerular no son reversibles y que los glomérulos afectados progresan a la esclerosis. Esto sugiere, que el pronóstico a largo plazo, va a depender de la extensión de la lesión renal durante el episodio agudo de la enfermedad. Numerosos estudios desde los iniciales de Habib (5) han confirmado que la MAT glomerular, que es el patrón histológico habitualmente descrito en la edad temprana en el SHU asociado con diarrea, tiene mucho mejor pronóstico que la MAT vascular (33,34). Se ha comprobado que cuando la lesión microangiopática afecta a menos del 50% de los glomérulos, la recuperación de la función renal puede ser completa. Por el contrario, la MAT arterial, que es más frecuente en niños mayores, en adultos y en las formas familiares del SHU/PTT, tiene muy mal pronóstico y más del 70% de los pacientes, evolucionan a la insuficiencia renal con hipertensión arterial (36,37). Por tanto, la intensa endarteritis proliferativa, es el factor que más va a determinar el pronóstico en un paciente con MAT. En general, los cambios glomerulares, por sí solos, no implican un mal pronóstico (38), excepto cuando se asocian a una extensa necrosis cortical.
Las lesiones de la MAT arterial son indistinguibles histológicamente de las lesiones vasculares de la esclerosis sistémica (ES) y de la nefroesclerosis maligna. La incidencia de afectación renal en la ES es difícil de precisar, pero la forma más grave de lesión renal, denominada «crisis renal esclerodérmica» (CRE), se caracteriza por el desarrollo de insuficiencia renal rápidamente progresiva (IRRP), generalmente asociada a proteinuria, hiperreninemia y anemia hemolítica microangiopática. Aunque esta forma agresiva de lesión renal suele acompañarse de hipertensión arterial maligna, la TA puede ser inicialmente normal o estar sólo ligeramente aumentada. Aproximadamente, el 20% de los pacientes con la forma difusa de ES, desarrollan la CRE, frente al 2% de los pacientes con la forma limitada de la enfermedad. En la CRE, las arteriolas, arterias interlobares y las arterias arcuatas más pequeñas, presentan cambios intensos, caracterizados por un engrosamiento fibromucoide concéntrico de la íntima, con importante reducción de la luz vascular (fig. 10). Pueden verse hematíes fragmentados en la intimal o atravesando la pared vascular y pequeños acúmulos de fibrina debajo del endotelio o subintimales. Las arteriolas pueden presentar trombos eosinófilos y áreas de necrosis fibrinoide, que se extienden al hilio glomérular. Las arterias de mayor calibre, presentan engrosamientos fibrosos inespecíficos de la íntima. Las alteraciones glomerulares son muy variables, predominando los cambios de tipo isquémicos.
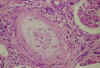
Fig. 10. Esclerosis
sistémica. Arteria interlobular con intenso engrosamiento fibromucoide de la
íntima (HE).
En la nefroesclerosis maligna, las lesiones afectan fundamentalmente a los glomérulos, arterias interlobulares y arteriolas. Las arteriolas presentan necrosis fibrinoide, asociada con trombos intraluminares que llegan a ocluir la luz. En la íntima y en la capa media, pueden verse hematíes fragmentados y en ocasiones infiltrados inflamatorios. La necrosis se puede extender a los glomérulos y asociarse con la formación de semilunas. Las arterias interlobulares también pueden presentar necrosis fibrinoide, pero la lesión más característica es el engrosamiento mucoide subintimal y la proliferación celular con disposición en capas concéntricas (fig. 11). En la íntima engrosada, pueden verse macrófagos de aspecto espumoso y la lamina elástica interna está reduplicada.
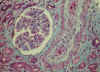
Fig. 11. Nefroesclerosis
maligna, inducida por cocaína, en mujer de 35 años. Se observa proliferación
fibromioide de la íntima con trombos de fibrina en la luz vascular.
Por tanto, las alteraciones vasculares descritas clásicamente en la ES y en la nefroesclerosis maligna, son idénticas a las lesiones de la MAT arterial. Además, los pacientes también presentan anemia hemolítica microangiopática, lo que origina que en muchos casos, no sea posible diferenciar la nefroesclerosis maligna del SHU/PTT del adulto. Por todo esto, en la actualidad, las lesiones renales que acompañan a la ES y a la nefroesclerosis maligna, se incluyen también en el grupo de las MAT (tabla I).

BIBLIOGRAFÍA
Moschcowitz E. An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capilaries: an undescribed disease. Arch Inter Med 1925; 36: 89-93.
Singer K, Bornstein FP, Wile SA. Thrombotic thrombocytopenic purpura: Hemorrhagic diathesis with generalized platelet thromboses. Blood 1947; 2: 542-54.
Symmers WSC. Thrombotic microangiopathic haemolytic anemia (thrombotic microangiopathy). Br Med J 1952; 2: 897-903.
Gasser C, Gautier E, Steck A, Siebenmann RE, Dechslin R. Hämolytisch-urämische syndrome bilaterale Nierenrindennekrosen bei akuten erworbenchen hämolytischen Anämien. Schweiz Med Wochenschr 1955; 85: 905-9.
Habib R, Mathieu H, Royer T. Maladie thrombotique artériolocapilaire du rein chez l’enfant. Rev Fr Etud Clin Biol 1958; 3: 891-4.
Remuzzi G, Ruggenenti P, Bertani T. Thrombotic Microangiopathy. En: Tisher CC, Brenner BM (eds): Renal Pathology: With clinical and functional correlations. Philadelphia, JB Lippincott. 1994 vol 2 p 1154-84.
Laszik L, Silva FG. Hemolytis-Uremic Syndrome, Thrombotic Thrombocytopenic Purpura and Systemic Sclerosis (Systemic Scleroderma). En: Jennette JC, Olson JL, Schwartz MM, Silva FG (eds): Heptinstall’ Pathology of the Kidney. Lippincott-Raven. 1998 vol 2 p 1003-57.
Ridolfi RL, Bell WR. Thrombotic thrombocytopenic purpura: Report of 25 cases and review of the literature. Medicine 1981; 60: 413-28.
Morel-Maroger L: Adult hemolytic-uremic syndrome. Kidney Int 1980; 18: 125-34.
Melnyk AM, Solez K, Kjellstrand CM. Adult hemolytic syndrome; a review of 37 cases. Arch Intern Med 1995; 155: 2077-84.
Robson WL, Leung AK, Montgomery MD. Causes of death in hemolytic-uremic syndrome. Child Nephrol Urol 1991; 11: 228-33.
Remuzzi G . HUS and TTP: Variable expresion of a single entity. Kidney Int 1987; 32: 292-308.
Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int 2001; 60: 831-46.
Remuzzi G, Ruggenenti P. The hemolytic uremic syndrome. Kidney Int 1995; 47: 2-19.
Baid S, Pascual M, Cosimi AB y cols. Virus and thrombotic microangiopathy. Transplantation 1999 Sep15; 68: 710-11.
Peraldi MN, Maslo C, Akposso K y cols. Acute renal failure in the course of HIV infection: a single-institution retrospective study of ninety-two patients and sixty renal biopsies. Nephrol Dial Transplant 1999; 14: 1578-85.
Abrahan B, Baud O, Bonnet E, Roger PM y cols .Thrombotic microangiopathy during HIV infection. A retrospective study performed in infectious diseases units in southern France. Presse Med 2001; 30: 581-5.
Stricker RB, Davis JA, Gershow J y cols. Thrombotic thrombocytopenic purpura complicating systemic lupus erytematus: case report and literature review from the plasmapheresis era [Review]. J Rheumatol 1992; 19: 1469-73.
Bridoux F, Vrtovsnik F, Noel C y cols. Renal thrombotic microangiopathy in systemic lupus erythematous: clinical correlations and long-term survival. Nephrol Dial Transplant 1998; 13: 298-304.
Musio F, Bohen EM, Yuan CM, Welch PG. Review of thrombotic thrombocytopenic purpura in the setting of systemic lupus erythematosus [Review]. Semin Arthritis Rheum 1998 Aug; 28: 1-19.
Sakaguchi S, Kitazama K, Watanabe M y cols. A case of primary antiphospholipid antibody syndrome with acute renal failure showing thrombotic microangiopathy. Am J Nephrol 1999; 19: 594-8.
Stefanidis I, Helmchen U, Schmitt M y cols. Coincidence of haemolytic uraemic syndrome and c-ANCA-associated rapidly progressive glomerulonephitis. Nephrol Dial Transplant 1998; 13: 1818-21.
Stave GM, Croker BP. Thrombotic mocroangiopathy in anti-glomerular basement membrane glomerulonephitis. Arch Pathol Lab Med 1984; 108: 747.
Kwaan HC, Gordon LI: Thrombotic microangiopathy in the cancer patient. Acta Haematol 2001; 106: 52-6.
Magee CC, Abraham K, Farrell J y cols. Renal thrombotic microangiopathy associated with interferon alpha treatment of chronic myeloid leukemia. Am J Kidney Dis 2000; 36: E5.
Young BA, Marsh CL, Alpers CE, Davis CL. Cyclosporine-associated thrombotic microangiopathy/hemolytic uremic syndrome following kidney and kidney-pancreas transplantation. Am J kidney Dis 1996; 28: 561-71.
Randhawa PS, Starzl TE and Demetris AJ. Tacrolimus (FK506)-associated renal pathology. Adv Anat Pathol 1997; 4: 265-76.
Zarifian A, Meleg-Smith S, O’Donovan R y cols. Cyclosporine-associated thrombotic microangiopathy in renal allografts. Kidney Int 1999; 55: 2457-66.
Burke GW, Cianco G, Cirocco R y cols. Microangiopathy in kidney and simultaneous pan creas/kidney recipients with tacrolimus: evidence of endothelin and cytokine involvement. Transplantation 1999; 68: 1336-42.
Pham PT, Peng A, Wilkinson AH y cols. Cyclosporine and tacrolimus-associated thrombotic microangiopathy. Am J Kidney Dis 2000; 36: 844-50.
Pichette V, Querin S, Schurch W y cols. Familial hemolitic-uremic syndrome and homozygous factor H deficiency. Am J Kidney Dis 1994; 24: 936-41.
Ohali M, Shalev H, Schlesinger M y cols. Hypocomplementemic autosomal recessive hemolytic uremic syndrome with decreased factor H. Pediatr Nephrol 1998; 12: 619-24.
Royer P, Habib R, Mathieu H. La microangiopathie thrombotique du rein chez l’enfant. Ann Pèdiat (Paris) 1960; 36: 572.
Habib R, Levy M, Gagnadoux MF y cols. Prognosis of the hemolytic uremic syndrome in children. Adv Nephol 1982; 11: 99-128.
Amir G, Rosenmann E, Drukker A. Acquired glomerulocystic kidney disease following haemolytic-uraemic syndrome. Pediatr Nephol 1995; 9: 614-6.
Morel-Maroger L, Kanfer A, Solez K, Sraer JD, Richet G. Prognostic importance of vascular lesions in acute renal failure with microangiopathic hemolytic anemia (hemolytic uremic syndrome): clinico-pathologic study in 20 adults. Kidney Int 1979; 15: 548-58.
Renaud C, Niaudet P, Gagnadoux MF, Broyer M, Habib R: Haemolytic uraemic syndrome: prognostic factors in children over 3 years of age. Pediatr Nephrol 1995; 9: 24-9.
Inward CD, Howie AJ, Fitzpatrick MM y cols.: Renal histopathology in fatal cases of diarrhoea-associated haemolytic uraemic syndrome. British Association for Paediatric Nephrology. Pediatr Nephrol 1997; 11: 556-9.
![]()