
Vol. 35, n.º 3, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 3, 2002 |
Eduardo Vázquez Martul, J. Veiga Barreiro
Servicio de Anatomía Patológica. Hospital «Juan Canalejo». 98 117 80 00. A Coruña. Carretera As Xubias, n.º 84, 15006 A Coruña
INTRODUCCIÓN
Desde que se realizó el primer trasplante renal en el Hospital Juan Canalejo, en el año 1982, se han llevado a cabo hasta el momento actual un total próximo a 1500 (tabla I). La experiencia acumulada ha permitido un mejor manejo clínico de los pacientes, en gran medida gracias al descubrimiento de potentes y eficaces fármacos inmunosupresores, entre los que debemos destacar la Cyclosporina (Cys) (1). Sin embargo, y a pesar del desarrollo de una amplia terapia inmunosupresora (2), el rechazo continúa siendo una de las principales causas de pérdida del injerto, que aproximadamente es de un 30% a los 5 años y que alcanza el 50% al cabo de los 10 años post-trasplante en programas de injertos de cadáver (3,4).

Desde un principio la biopsia renal sigue siendo el mejor método para diagnosticar la existencia de rechazo, principal objetivo de la biopsia, (tabla II) y en caso positivo, la indicación de un tratamiento inmunosupresor adecuado (5). Las sucesivas reuniones de Banff (6,7), han intentado establecer criterios diagnósticos histopatológicos objetivos, y graduación de la intensidad de las lesiones renales con triple finalidad: diagnóstica, correlación clínico-patológica y establecer un pronóstico (8-13).

Sin embargo, para una correcta valoración de la biopsia del injerto renal, es altamente recomendable una estrecha relación clínico-patológica, con un conocimiento previo de aquellos factores que pueden influir en las características histopatológicas, tanto en el período inicial como en períodos más crónicos (tabla III). Entre estos factores debemos destacar aquellos relacionados con el donante (14-16), factores relacionados con la calidad de preservación del injerto y tiempos de isquemia fría (17-19), factores dependientes del protocolo inmunosupresor (20-21), y por supuesto aquellos relacionados con el receptor que puedan influir en la función del injerto, tanto en los periodos iniciales como tar díos (22), destacando aquellas enfermedades susceptibles de recidiva (23,24).

También es muy importante conocer los principales mecanismos fisiopatológicos, inmunológicos y no inmunológicos, que faciliten valorar el substrato histopatológico tanto en la fase aguda (25), como crónica de la evolución (26-28). En este contexto, la participación del patólogo es fundamental, no sólo para una adecuada monitorización terapéutica, limitando la morbilidad y costo del programa, sino colaborando en la mejor supervivencia del injerto y del paciente.
PRINCIPALES ESTADÍOS CLÍNICOS Y MECANISMOS FISIOPATOLÓGICOS
En la correlación clínico patológica y a lo largo de la evolución del trasplante, se dan unos momentos evolutivos en los que la mala función renal del injerto tiende a estar asociada con determinados mecanismos fisiopatológicos, inmunológicos y no inmunológicos, que es necesario conocer para valorar el substrato histopatológico que hegemoniza un determinado período clínico (tabla IV). Existen cuatro períodos clínicos interrelacionados que tienen importancia en la valoración de la biopsia, en la correcta monitorización terapéutica y por consiguiente, en la supervivencia del injerto y del paciente.
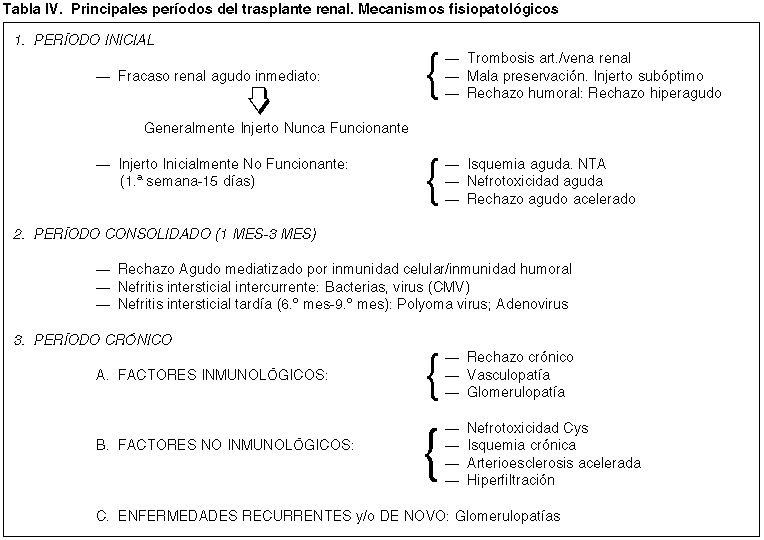
PERÍODO INICIAL. INJERTO NUNCA FUNCIONANTE (INF) E INJERTO INICIALMENTE NO FUNCIONANTE (IINF)
— Factores relacionados con Injerto Nunca Funcionante (INF). Son factores que implican una ausencia de función inicial del injerto, y casi siempre conlleva mal pronóstico y pérdida del mismo. En una revisión de nuestros pacientes trasplantados (17,29,30), el 11% de un total de 747 pacientes revisados, fueron etiquetados de (INF). Las principales causas no inmunológicas se atribuyen a trombosis del pedículo vascular, no función primaria, generalmente por mala preservación y/o técnicas quirúrgicas, y ocasionalmente ruptura de injerto (29-31). Entre las causas con base inmunológica debemos citar el Rechazo Hiperagudo (RH), de presentación inmediata y consecuencia de la producción masiva de anticuerpos preformados, actualmente muy ocasional debido al conocimiento previo del panel de anticuerpos tanto del donante como del receptor. Sin embargo es confuso el conocimiento del papel de actuación y los efectos de otros mecanismos inmunológicos en esta fase inmediata al margen del RH (32,33).
— Injerto Inicialmente No Funcionante (IINF). Se define como aquel injerto que precisa hemodiálisis durante la primera semana (20). La incidencia aproximada en nuestra estadística es de un 37% de casos (29). El principal mecanismo fisiopatológico de insuficiencia renal está relacionado con la isquemia aguda y el substrato histopatológico corresponde a una Necrosis Tubular Aguda (NTA) (34,35).
— Los factores que influyen en una insuficiente función inicial del injerto son múltiples y su relación con el pronóstico muy discutidos. La edad del donante, es un factor destacable sobre todo en donantes añosos por encima de 65 años de edad, con implicaciones no sólo en la mala función inicial sino también en el pronóstico a largo plazo del injerto (14,30,36,37).
Existen otros factores, al margen de la edad, causantes de IINF y entre los que destacaremos: donantes fallecidos en asistolia, pacientes diabéticos, hipertensos y fallecidos en accidente cerebro-vascular (ACV) (15,38). Este tipo de donantes, antiguamente descartados, hoy día son aceptados como injertos válidos, debido a la presión que ejerce la extensa lista de espera de pacientes en programa de insuficiencia renal crónica, y en parte a los buenos resultados obtenidos incluso de tipo económico, en comparación con los gastos y pronóstico de otros pacientes sometidos a diálisis crónica (37-39).
Es muy importante conocer todos estos datos con el fin de una adecuada valoración de las posibles lesiones glomerulares, túbulo-intersticiales y vasculares existentes en la primera biopsia del injerto ya que la presencia de un porcentaje de glomeruloesclerosis superior al 20%, intensa arterioesclerosis y sobre todo hialinosis arteriolar, repercutirán negativamente en la función inicial del injerto (16,40-42). En algunos estudios, sólo el 38% de donantes de cadáveres muestran una biopsia completamente normal (5).
— Factores relacionados con la mala preservación y en relación directa con tiempos de isquemia fría, superiores a 24 horas (17,19), también tienen una repercusión negativa, causando un incremento de NTA inicial, dificultando el estudio de la biopsia, y por consiguiente el manejo clínico del paciente sobre todo en aquellos casos con mala función inicial prolongada superior a la primera semana (30).
Dentro de esta primera fase cronológica del injerto, que ocupa los primeros días, debemos citar las consecuencias negativas de la terapia inmunosupresora, concretamente la Nefrotoxicidad con Cys y Tacrolimus (43). Estos inmunosupresores ejercen una importante vasoconstricción mantenida, relacionada con la acción sobre el endotelio y secreción de endotelinas, que potencian el daño isquémico tisular y facilitan la NTA (44,45).
En este periodo inicial, junto con estos mecanismos previos no inmunológicos consecuentes de la isquemia aguda renal, pueden existir mecanismos inmunológicos no bien conocidos de Rechazo Agudo (RA), generalmente denominado Rechazo Agudo Acelerado(RAA), en la mayoría de los casos mediatizado por la formación de anticuerpos que constituyen un ejemplo de Rechazo Humoral (RH) (45). El RAA, ocurre con mas frecuencia en pacientes sensibilizados por trasplantes previos y conlleva un mal pronóstico con una alta tasa de pérdida del injerto (46,47).
PERÍODO CONSOLIDADO. FRACASO RENAL RELACIONADO CON RECHAZO AGUDO (RA)
El RA sigue hegemonizando la principal patología del trasplante renal y es el factor de mayor impacto relacionado con el Rechazo Crónico (RC) y la pérdida definitiva del injerto (17,19,30,48) (tabla V). La revisión de factores relacionados con la supervivencia del injerto, teniendo en cuenta variables como donantes en asistolia, tiempo de espera, isquemia fría, incompatibilidad HLA-DR, y NTA, el RA resultó el factor de mayor impacto (17) (tablaV). Entre un 70-80% de episodios de RA acontece durante los primeros tres meses postrasplante, sin embargo puede ocurrir muy precozmente en los primeros días o más tardíamente después del sexto mes. Esta cronología precoz o tardía parece tener relación con el pronóstico y el RC (24,47,48).
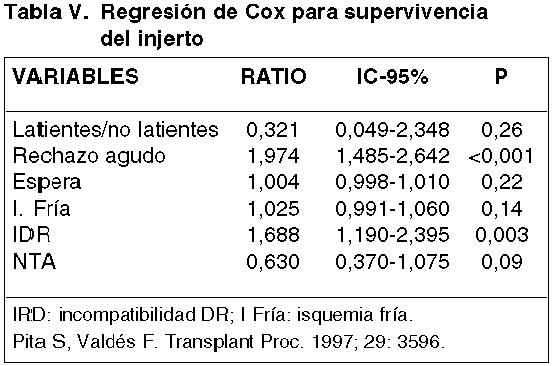
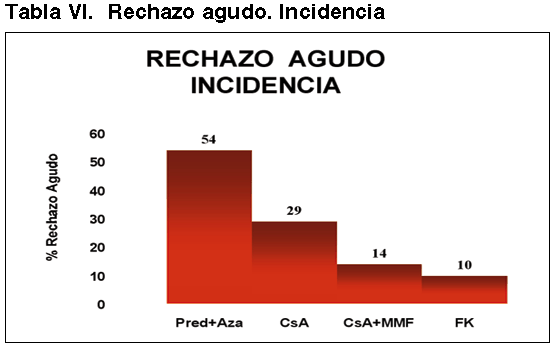
El mecanismo fisiopatológico está relacionado con la respuesta inmunitaria de tipo celular, que se manifiesta histológicamente por la infiltración túbulo-intersticial y vascular por población linfocitaria preferemente T, y cuyos criterios histológicos han sido descritos en las diferentes reuniones de Banff (6,7). En nuestra experiencia, el 35% de pacientes presentaron al menos una crisis de RA después del trasplante renal, la mayoría, entre la segunda y la cuarta semana de la evolución post-trasplante (29). La incidencia de RA ha disminuido notablemente y a lo largo de la historia con una estrecha relación con el tipo de protocolo inmunosupresor, variando desde un 50% en la época de utilización de la Azatioprina, hasta un 10% actual después de la introducción del tacrolimus (FK) (tabla VI).
En cualquier momento de la evolución, pero siempre muy relacionado con el diagnóstico diferencial del RA, se debe considerar como causa de insuficiencia renal, una nefritis intersticial de causa infecciosa. La terapia inmunosupresora predispone a infecciones intercurrentes, bien sea por causa bacteriana, pero más frecuentemente de etiología vírica entre las que destacamos CMV (49,50), polyoma-virus (51), adenovirus (52), y Ebstein Barr-virus (EBV) (49,53,54).
PERÍODO CRÓNICO. FRACASO CRÓNICO DEL INJERTO(FCI)
El deterioro progresivo de la función renal, asociado generalmente a proteinuria e hipertensión es una de las principales características del FCI (26,28,55,56). El cuadro histopatológico es muy heterogéneo (56,57), como heterogéneas son las causas relacionadas con la insuficiencia crónica del injerto (55). En este período existen también dos mecanismos fisiopatológicos:
— Un mecanismo inmunológico representado por Rechazo Crónico (RC) propiamente dicho y que se caracteriza por vasculopatía del RC que puede asociarse infrecuentemente a una glomerulopatía post-trasplante (27,56-58).
— Otro mecanismo, no inmunológico, hegemonizado por la isquemia y la nefrotoxicidad crónica (26,43,56,57) con fibrosis intersticial y lesiones crónicas arteriolares y arteriales tipo arterioesclerosis a veces difíciles de diferenciar de las propias producidas por RC.
También dentro de este período de FCI, podemos incluir a gran parte de enfermedades glomerulares recurrentes y/o glomerulopatías de novo que normalmente se asocian a un cuadro proteinúrico florido o un síndrome nefrótico (24,59). Alguna glomerulopatía recurrente, como la esclerosis-hialinosis focal segmentaria (24) y el síndrome hemolítico-urémico (SHU) (23) pueden presentarse de forma muy precoz con mal pronóstico y pérdida del injerto.
La subdivisión de diferentes etapas clínicas, debemos aceptarlo como un esquema o guía de ayuda en la valoración de la biopsia renal, y nunca entenderlo como situaciones clínico- patológicas independientes, ya que no podemos olvidar que puede acontecer una crisis de RA, celular o humoral en cualquier momento evolutivo y a su vez los mecanismos inmunológicos y no inmunológicos estar interrelacionados. También debemos tener en cuenta otros procesos intercurrentes, muy relacionados con el estado de inmunodepresión del paciente, como nefritis intersticiales de origen infeccioso bacteriano o vírico, que dificultan el diagnóstico diferencial con el RA.
SUBSTRATO HISTOPATOLÓGICO DEL INJERTO NUNCA FUNCIONANTE (INF)
Dentro de los mecanismos no inmunológicos causantes de la no función del injerto, debemos citar la trombosis del pedículo vascular renal, que puede alcanzar hasta un 30% de las causas de no función (29). El diagnóstico es radiológico y el estudio macroscópico e histopatológico de la pieza de nefrectomía, demostrará trombosis arterial y/o venosa con infarto, generalmente hemorrágico con intensa congestión capilar glomerular y signos de necrosis tubular. Otra causa, también importante sobre todo en los primeros períodos preciclosporina, era la rotura renal en gran parte debido al intenso edema intersticial, observándose 24 casos de un total de 745 pacientes revisados de nuestra casuística (29).
Entre los mecanismos inmunológicos causante de la ausencia de función del injerto, debemos citar el RH, prácticamente inexistente en la actualidad. Este tipo de rechazo inmediato, es el prototipo de rechazo humoral, y ocurre cuando un riñón es trasplantado en un receptor presensibilizado ante el panel de anticuerpos del donante y por consiguiente ha preformado anticuerpos circulantes contra antígenos generalmente del tipo anti-HLA del donante (60,61). El resultado es una inmediata lesión del endotelio con trombosis masiva e irreversible. El cuadro puede ser lo suficientemente intenso para producir infartos y hemorragias intersticiales con un diagnóstico diferencial con trombosis no inmunológica del injerto. En estudios con inmunofluorescencia pueden existir depósitos capilares y en pequeñas arterias y arteriolas de IgM, IgG, fibrina y C3 (5,62).
SUBSTRATO HISTOPATOLÓGICO DEL INJERTO INICIALMENTE NO FUNCIONANTE (IINF)
En nuestra experiencia y después de casi 1.500 pacientes trasplantados, el IINF es una de las principales causas de biopsias reiteradas y de morbilidad coincidiendo con la experiencia de otros autores (14,29,30,63). La instauración de tratamiento con Cys, la aceptación de injertos de cadáver de edades superiores a 65 años, y pacientes fallecidos en asistolia, ha potenciado el porcentaje de IINF que se acerca actualmente a un 30% (14,30,63). Los riñones de donantes con asistolia o de fallecidos en accidentes cerebro-vascular, así como aquellos con isquemia fría superior a 24 horas, son subsidiarios, no solo de mala función inicial, sino de mal pronóstico del injerto con mayor porcentaje de rechazos agudos (36-38,63,64).
El sustrato histopatológico del IINF está hegemonizado por un cuadro de NTA. Uno de los principales motivos de controversia en la literatura, está relacionado con el impacto de la NTA en el pronóstico del injerto a corto y largo plazo (14,36,63). En una revisión propia de 145 biopsias de injerto inicialmente no funcionante, el 25% de las mismas tenían una NTA no complicada con otros procesos (65).
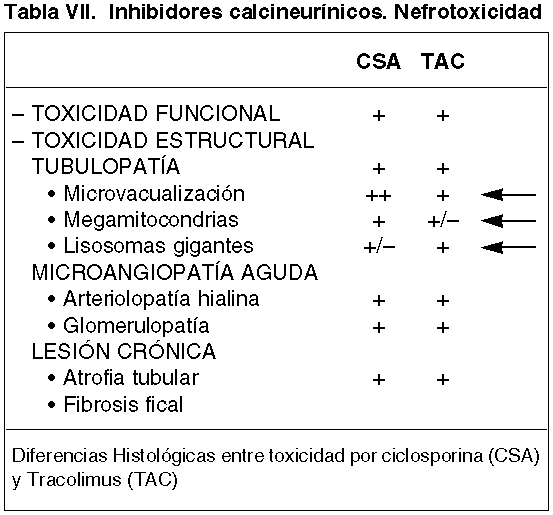
El cuadro de NTA se caracteriza clásicamente por la presencia de abundante descamación de células tubulares, presencia de cilindros fuchinófilos con las tinciones de tricrómico de Marson, dilatación de túbulos con fragmentación de la membrana basal tubular, presencia de mitosis y prominentes nucleolos (fig. 1a). La pérdida de las microvellosidades apicales de los túbulos proximales, el aplanamiento del laberinto basal-lateral y la alteración mitocondrial sobre todo de la pars recta de los túbulos proximales, junto con la aparición de acentuaciones filamentosas de actina, son alteraciones ultraestructurales muy características de cualquier NTA (fig. 1b) (66-68). Algunos autores observan diferencias entre la NTA, existente en la primera fase del IINF, y la NTA que ocurre en otro contexto al margen del trasplante (68). Las principales diferencias observadas por Olsen (68), se cifra en menor afectación del reborde en cepillo, más cantidad de depósitos de calcio intratubular, mayor grado de componente inflamatorio intersticial y de descamación celular. Es muy posible que las características morfológicas de la NTA post-trasplante sean cualitativa y cuantitativamente diferentes en pacientes inmunodeprimidos con Cyclosporina, agente que produce y potencia la incidencia de IINF (44).
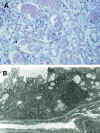
Fig. 1. a) Intensa descamación
celular intratubular. b) Estudio ultraestructural con pérdida de
microvellosidades apicales (flecha superior), aplanamiento del laberinto basal
con placas densas (flechas inferiores) y lesión mitocondrial.
El principal problema en la valoración de la biopsia renal en esta primera fase, surge en determinar si el daño es exclusivamente isquémico, si existe patología concomitante secundaria a toxicidad por CyS, y lo que es más importante si existe un RA precoz sobre todo en aquellos casos de IINF tardíos o prolongados, en los que se inicia un componente inflamatorio intersticial, generalmente por macrófagos, con ausencia de «tubulitis» (64). En definitiva, el problema más arduo está en poder descartar una NTA «pura» y de tipo isquémico, o una NTA «complicada» con un proceso tóxico, inflamatorio o por la existencia de un RA. En nuestro experiencia, tras revisión de 148 biopsias de INF el 25% correspondían a NTA isquémica, el 21% a lesiones compatibles con toxicidad por Cys, y en el 18% la NTA se asociaba a componente inflamatorio, generalmente macrófagos sin tubulitis, en el que se plantea un diagnóstico diferencial con lesiones de RA precoz y/o toxicidad por CyS (65). En estos casos es útil el estudio inmunohistoquímico ya que en el RA debe existir un predominio de linfocitos T si bien la presencia de macrófagos no descarta la presencia de una crisis de RA (5).
Dentro del sustrato histopatológico del IINF, debemos citar las lesiones producidas por nefrotoxicidad, preferentemente Cys y Tacrolimus. Las lesiones histopatológicas producidas por estos inhibidores calcineurínicos, son ampliamente descritas en la literatura (43-45), no existiendo ningún criterio histopatológico específico que indique nefrotoxicidad por Cys ni tampoco diferencias histopatológicas entre CyS y Tacrolimus (43) (tabla VII). Las lesiones se definen de dos tipos:
a) Nefrotoxicidad funcional, consecuencia de un vaso-espasmo mantenido, en la que se puede evidenciar una oclusión de luces arteriolares, vacuolización y necrosis de miocitos de arteriolas y de pequeñas arterias. En estos casos el problema diagnóstico consiste en confundir la eosinofilia de la necrosis del miocito con una veradera necrosis fibrinoide mediatizada por anticuerpos propio de un Rechazo humoral
b) Nefrotoxicidad tubular propiamente dicha, cuyo rasgo más característico es la microvacuolización isométrica de túbulos proximales, presencia de megamitocondrias, y dilatación de capilares peritubulares (figs. 2a y b).
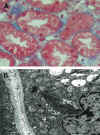
Fig. 2. a) Nefrotoxicidad.
Microvacuolización. b) Estudio ultraestructural. Megamitocondrias.
También se han descrito lesiones tipo microangiopáticas a nivel glomerular muy similares a las descritas en el SHU (43). Es muy posible que esta lesión no sea nefrotóxica «per se», sino el producto de un verdadero síndrome hemolítico urémico leve, con representación histológica muy segmentaria, desencadenado por la intensa lesión de las células endoteliales producidas por la Cys.
En este período de mala función inicial del injerto, debemos considerar al Rechazo Agudo precoz, sobre todo en aquellos casos de IINF muy prologados, en los que no solamente se observa NTA sino también componente inflamatorio acompañante. En estos casos es útil la técnica del PAS para determinar con exactitud la existencia de tubulitis. El diagnóstico de RA de leve intensidad en estos períodos es quizás el diagnóstico más difícil y complicado. Es precisamente esta dificultad la que lleva a admitir el término de Lesión borderline (6,7,9,69). En resumen, las consecuencias del IINF consisten en una disminución de la supervivencia, predisposición a RA y un incremento de las dificultades en el manejo terapéutico que conlleva un aumento de la morbilidad y, lógicamente del costo por paciente y trasplante.
RECHAZO AGUDO (RA). SUSTRATO HISTOPATOLÓGICO
El principal objetivo de la biopsia del injerto renal es, sin duda alguna, el diagnóstico de rechazo. El RA, bien sea precoz o tardío, no sólo facilita el rechazo crónico, sino que es la principal causa de pérdida del injerto (17,24,47,48).
En una revisión parcial de la casuística de nuestro Hospital (29), el 35% de los pacientes presentaron al menos una crisis de RA después del trasplante renal. En 173 casos, (es decir el 23,7%), la primera crisis de rechazo agudo se diagnosticó después de las dos primeras semanas de evolución y en 52 casos (7%), se produjo entre la tercera y cuarta semana, y sólo en 16 casos (2,4%) la primera crisis se produjo después del tercer mes. En el 70% de los casos el diagnóstico se basó en la biopsia, siendo el patrón histológico predominante el rechazo túbulo-intersticial agudo (29).
Es importante para un correcto diagnóstico de RA, por medio de la biopsia renal, que ésta tenga un alto grado de sensibilidad y reproducibilidad, basando el diagnóstico en los principales mecanismos patogénicos implicados en el rechazo. Clásicamente se han descrito dos mecanismos inmunológicos: a) Rechazo humoral, secundario a la formación de anticuerpos y complejos inmunocirculantes; y b) Rechazo celular, conocido como rechazo túbulo intersticial agudo (RTIA), más frecuente y consecuencia de la estimulación de la respuesta T linfocitaria a CD8 y CD4 (5).
RECHAZO TÚBULO-INTERSTICIAL AGUDO (RTIA)
La principal característica es la presencia de componente mononuclear a nivel intersticial, con infiltración del epitelio tubular (tubulitis), endotelio vascular (endotelialitis) y glomerular. Puede acompañarse, aunque no necesariamente de diferente grado de edema y focos de hemorragia. El principal componente celular inflamatorio es linfocitario, CD8 y CD4, pero pueden identificarse macrófagos, eosinófilos, mastocitos así como celularidad plasmática (5,70-72).
Admitir en que medida las características histopatológicas son específicas y sensibles para hacer un diagnóstico de RTIA, y de qué manera están relacionadas con el pronóstico, ha sido la principal discusión a lo largo de la literatura (5-13,69,73). Con el objeto de solucionar este problema, diferentes grupos de trabajo han propuesto criterios diagnósticos, así como grados de intensidad. La dualidad existente entre la clasificación propuesta por la CCTT (Cooperativa Clinical Trials Trasplantation) (73), y el grupo de Banff (6) se ha unificado recientemente (7) y confirmado en la última reunión celebrada en el 2001 en Banff.
Básicamente se acepta graduar la tubulitis (presencia de células mononucleares en el epitelio tubular) y endotelialitis (infiltración subendotelial), como los dos aspectos histopatológicos básicos en el diagnóstico y pronóstico del rechazo (7,73).
La tubulitis, en relación con el número de células mononucleares que infiltran el epitelio tubular se gradúa de 0 a 3 (7) (fig. 3)
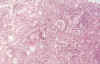
Fig. 3. RTIA. Denso infiltrado
tubular con tubulitis.
T-1: menos de 4 células mononucleares por sección tubular.
T-2: focos de 5 a 10 células y,
T-3: más de 10 células, o al menos presencia de dos áreas en donde exista destrucción de la membrana basal tubular, acompañada de moderado componente intersticial y más de 5 células monocitarias por sección tubular en cualquier parte de la biopsia.
La endotelialitis, también es graduada de 0 hasta 3, dependiendo de su intensidad y extensión, considerándose el grado más severo cuando existe arteritis trasmural y/o necrosis fibrinoide (7) (fig. 4).
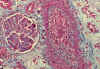
Fig. 4. Endotelialitis en vaso
arterial.
En relación con la valoración de la biopsia renal, no parece existir dificultad alguna en aquellos grados intensos de rechazo (I/B), caracterizada por afectación de más del 25% del área intersticial y con más de 10 células monocitarias por sección tubular (7). Tampoco es complicada la identificación de rechazos, grados II/B y grado III, caracterizadas por intensa endotelialitis (II/B) con arteritis trasmural (grado III). Existe unanimidad en la literatura que casos de afectación arterial (II/B y III), conlleva peor pronóstico sobre todo si se evidencia necrosis fibrinoide (8,9,74). También se ha descrito que rechazos con una tubulitis severa (t-3, grado I/B), el pronóstico es peor que rechazos leves grado I/A (11).
Contrariamente a lo que ocurre con los rechazos agudos con manifiesta representación histológica (I/B, II/A, II/B y III), en los rechazos leves (IA) y sobre todo en las lesiones borderline, el grado de reproducibilidad histológica entre autores es mucho más problemática (57,73). Este hecho puede explicar las discrepancias existentes en la literatura entre rechazos borderline y respuesta al tratamiento. En buena lógica, una afectación leve renal debería responder al tratamiento en un 100% de los casos (9), sin embargo, este hecho no se constata en otras series con respuestas completas en sólo un 43% (75), e incluso no respuesta en un 25% (69). Es muy posible que en estos casos puedan participar otros factores, entre los que debemos destacar, junto con la calidad y no representatividad de la biopsia, lesiones de tipo inflamatorio diferentes al RA.
Toda biopsia con componente inflamatorio túbulo-intersticial sin clara tubulitis, debe obligar a un diagnóstico diferencial con procesos inflamatorios (76) entre los que se destaca la infección viral por CMV (48,49), virus-polyoma tipo BK (51), adenovirus (52), y virus de Ebstein-Barr (53,54). El tipo de celularidad inflamatoria también tiene importancia ya que se ha propuesto que la población de macrófagos y celularidad plasmática puede tener un efecto negativo sobre función del injerto renal y ser un cofactor del rechazo crónico y peor respuesta al tratamiento (5,71,72). Un diagnóstico diferencial difícil se plantea entre RA e infección por virus tipo polyoma o BK, en donde puede existir un intenso componente inflamatorio linfocitario e incluso tubulitis (77). En estos casos la presencia de células tubulares con cambios citoplásmicos y nucleares de tipo cariomegálico, hipercromasia, apoptosis y anisocoriosis puede orientar hacia el diagnóstico que se debe confirmar con microscopio electrónico y utilización del anticuerpo SV40 (51,77).
En resumen, diremos que se da una buena sensibilidad y reproducibilidad histológica, así como relación clínico-pronóstica en todos los casos en donde existe afectación severa túbulo-intersticial y daño vascular (11,13). En aquellos casos en los que la lesión histopatológica sea de grado leve, o en los que no existe una correlación con buena respuesta al tratamiento, lo lógico es pensar que existan otros hechos patológicos concomitantes.
Algunos autores reconocen como, Glomerulopatía aguda del rechazo o Rechazo Agudo Glomerular a una lesión glomerular caracterizada por hipercelularidad, hiperplasia de células endoteliales e infiltración de células mononucleares CD4+,CD8+ y macrófagos (5,78,79). Este tipo de afectación glomerular ocurre infrecuentemente en menos del 7% de biopsias, es un factor de peor pronóstico (5) y se ha discutido su asociación con infección por CMV (80). Para otros autores es una forma inicial de glomerulopatía crónica o Glomerulopatía del trasplante no mediatizada por anticuerpos y de etiopatogenia desconocida (81-83).
RECHAZO AGUDO HUMORAL (RAH)
El rechazo renal mediado por la formación de anticuerpos citotóxicos antidonante, generalmente contra antígenos HLA, clase I y raramente clase II, es el principal mecanismo de rechazo humoral. Puede acontecer, bien de forma inmediata tras el trasplante, conocido como Rechazo Hiperagudo (RH), previamente comentado, o de forma más tardía, y a lo largo de los primeros meses, conocido también como Rechazo vascular acelerado, muy relacionado con pacientes retrasplantados. Anticuerpos citotóxicos circulantes de clase I, han sido detectados hasta en un 23-38% de pacientes trasplantados (60-63). El pronóstico es pobre, ya que más del 50% pierden definitivamente la función renal (63). En nuestra experiencia la incidencia es baja, aproximadamente del 4% y hasta el momento muy asociado a pacientes retrasplantados e hipersensibilizados (comunicación personal).
Las características histopatológicas son variables, habiéndose descritos casos con muy poca expresión morfológica, limitándose a edema intersticial y aislados signos de NTA, casos de afectación moderada con marginación de leucocitos en los capilares peritubulares y congestión capilar, y por último, casos muy floridos, en los que se observan trombos en capilares peritubulares y glomerulares, lesiones tipo MAT y necrosis fibrinoide arterial y arteriolar (62,84). El estudio con IF revela depósitos de IgG, IgM, C3 y fibrina (figs. 5a y b). Muy recientemente se han descrito depósitos de C4d en los capilares peritubulares como un marcador muy característico de rechazo humoral (85).
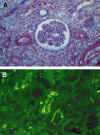
Fig. 5. a) Rechazo humoral.
Necrosis fibrinoide en arteria. b) Inmunofluorescencia directa con depósitos de
C3, IgM, IgG y fibrina.
Sin embargo, el diagnóstico diferencial de RAH, con inicio de trombosis capilar, puede plantear un diagnóstico histológico diferencial difícil con procesos que pueden cursar con lesiones tipo MAT. Entre estos cuadros debemos destacar la presencia de infecciones virales (86), crioglutininas y virus C en el período precoz post-trasplante (87), infecciones por CMV (88), SHU recurrente (23), así como pacientes con tratamiento de OKT3 que facilita los fenómenos de trombosis (89).
FRACASO CRÓNICO
La extensión del tema, nos impide describir con detalle el substrato histopatológico del Fracaso Crónico del Injerto, en donde , al igual que hemos descrito en el periodo inicial con el INF, participan fundamentalmente dos mecanismos fisiopatológicos (tabla VIII):
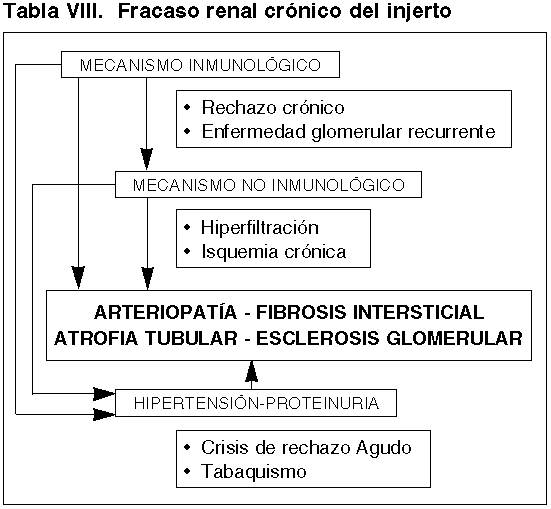
a) No inmunológicos, liderados por el efecto de la isquemia crónica, con producción de esclerosis vascular y glomerular, fibrosis intersticial con reducción de masa renal funcionante que ponen en marcha, cerrando un circulo vicioso, mecanismos de hiperfiltración renal (26,57).
b) Mecanismos inmunológicos cuyo sustrato más relevante y representativo es la Vasculopatía de Rechazo Crónico y la Glomerulopatía del Trasplante (5,27,35,56,57,58, 81,82,83) (figs. 6a y b).
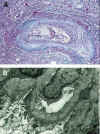
Fig. 6. a) Vasculopatía del
rechazo crónico con intensa endotelialitis. b) Glomerulopatía del trasplante.
Estudio ultraestructural con laminación de membrana basal glomerular.
La discusión de la complicada patogenia, su histopatología, glomerular, vascular y túbulo-intersticial, su posible relación con procesos metabólicos de alteración lipídica que la relacionan con la arterioesclerosis y ateromatosis (26), procesos víricos e infección por CMV(49), así como clasificación y graduación (10) aconsejan un capítulo independiente.
BIBLIOGRAFÍA
Kahan BD. Cyclosporine. N Engl J Med. 1989; 321: 1725-38.
Valdés F. Nuevos inmunosupresores en trasplante renal. Nefrología 1997; 17: 73-84.
Montagnino G, Tarantino A, Maccario M, Elli A, Cesana B, Ponticelli C. Long-term results with cyclosporine monotherapy in renal transplant patients: a multivariate analysis of risk factors. Am J Kidney Dis 2000; 35:1135-43.
Gjertson DW, Terasaki PI, Takemoto S, Mickey MR. National allocation of cadaveric kidneys by HLA matching. Projected effect on outcome and costs. N Engl J Med 1991; 324: 1032-6.
Colvin RB. The renal allograft biopsy. Kidney Int 1996; 50: 1069-82.
Racusen LC, Solez K, Colvin RB, Bonsib SM, Castro MC, Cavallo T, Croker BP, Demetris AJ, Drachenberg CB, Fogo AB, Furness P, Gaber LW, Gibson IW, Glotz D, Goldberg JC, Grande J, Halloran PF, Hansen HE, Hartley B, Hayry PJ, Hill CM, Hoffman EO, Hunsicker LG, Lindblad AS, Yamaguchi Y, et al. The Banff 97 working classification of renal allograft pathology. Kidney Int 1999; 55: 713-23.
Solez K, Benediktsson H, Cavallo T, Croker B, Demetris AJ, Drachenberg C, Emancipator S, Furness PN, Gaber LW, Gibson IW, Gough J, Gupta R, Halloran P, Hayry P, Kashgarian M, Marcussen N, Massy ZA, Mihatsch MJ, Morozumi K, Noronha I, Olsen S, Papadimitriou J, Paul LC, Picken M, Racusen LC, et al. Report of the Third Banff Conference on Allograft Pathology (July 20-24, 1995) on classification and lesion scoring in renal allograft pathology. Transplant Proc 1996; 28: 441-4.
Mueller A, Schnuelle P, Waldherr R, van der Woude FJ. Impact of the Banff '97 classification for histological diagnosis of rejection on clinical outcome and renal function parameters after kidney transplantation. Transplantation 2000; 69: 1123-7.
Gaber LW, Moore LW, Alloway RR, Flax SD, Shokouh-Amiri MH, Schroder T, Gaber AO. Correlation between Banff classification, acute renal rejection scores and reversal of rejection. Kidney Int 1996; 49: 481-7.
Dimeny E, Wahlberg J, Larsson E, Fellstrom B. Can histopathological findings in early renal allograft biopsies identify patients at risk for chronic vascular rejection? Clin Transplant 1995; 9: 79-84.
Minervini MI, Torbenson M, Scantlebury V, Vivas C, Jordan M, Shapiro R, Randhawa PS. Acute renal allograft rejection with severe tubulitis (Banff 1997 grade IB). Am J Surg Pathol 2000; 24: 553-8.
Solez K, Hansen HE, Kornerup HJ, Madsen S, Sorensen AW, Pedersen EB, Marcussen N, Benediktsson H, Racusen LC, Olsen S. Clinical validation and reproducibility of the Banff schema for renal allograft pathology. Transplant Proc 1995; 27: 1009-11
Kooijmans-Coutinho MF, Hermans J, Schrama E, Ringers J, Daha MR, Bruijn JA, van der Woude FJ. Interstitial rejection, vascular rejection, and diffuse thrombosis of renal allografts. Predisposing factors, histology, immunohistochemistry, and relation to outcome. Transplantation 1996; 61: 1338-44.
Koning OH, Ploeg RJ, van Bockel JH, Groenewegen M, van der Woude FJ, Persijn GG, Hermans J. Risk factors for delayed graft function in cadaveric kidney transplantation: a prospective study of renal function and graft survival after preservation with University of Wisconsin solution in multi-organ donors. European Multicenter Study Group. Transplantation 1997; 63: 1620-8.
Tullius SG, Volk HD, Neuhaus P. Transplantation of organs from marginal donors1. Transplantation 2001; 72: 1341-9.
Karpinski J, Lajoie G, Cattran D, Fenton S, Zaltzman J, Cardella C, Cole E. Outcome of kidney transplantation from high-risk donors is determined by both structure and function. Transplantation 1999; 67: 1162-7.
Pita S, Valdes F, Alonso A, Rivera CF, Cao M, Fontan MP, Carmona AR, Mongalian J, Adeva A, Lorenzo D, Oliver J. The role of cold ischemia on graft survival in recipients of renal transplants. Transplant Proc 1997; 29: 3596-7.
Alexander JW, Bennett LE, Breen TJ. Effect of donor age on outcome of kidney transplantation. A two-year analysis of transplants reported to the United Network for Organ Sharing Registry. Transplantation 1994; 57: 871-6.
Asderakis A, Dyer P, Augustine T, Worthington J, Campbell B, Johnson RW. Effect of cold ischemic time and HLA matching in kidneys coming from «young» and «old» donors: do not leave for tomorrow what you can do tonight. Transplantation 2001; 72: 674-8.
Halloran PF, Aprile MA, Farewell V, Ludwin D, Smith EK, Tsai SY, Bear RA, Cole EH, Fenton SS, Cattran DC. Early function as the principal correlate of graft survival. A multivariate analysis of 200 cadaveric renal transplants treated with a protocol incorporating antilymphocyte globulin and cyclosporine. Transplantation 1988; 46: 223-8.
Rush DN, Jeffery JR, Gough J. Protocol biopsies in stable renal transplant patients under triple immunosuppression: results at 6 months. Transplant Proc 1994; 26: 2576.
Perez Fontan M, Rodríguez-Carmona A, Bouza P, Garcia Falcon T, Adeva M, Valdes F, Oliver J. Delayed graft function after renal transplantation in patients undergoing peritoneal dialysis and hemodialysis. Adv Perit Dial 1996; 12: 101-4.
Miller RB, Burke BA, Schmidt WJ, Gillingham KJ, Matas AJ, Mauer M, Kashtan CE. Recurrence of haemolytic-uraemic syndrome in renal transplants: a single-centre report. Nephrol Dial Transplant 1997; 12: 1425-30.
Cameron JS. Glomerulonephritis in renal transplants. Transplantation 1982; 34: 237-45.
Cosio FG, Pelletier RP, Falkenhain ME, Henry ML, Elkhammas EA, Davies EA, Bumgardner GL, Ferguson RM. Impact of acute rejection and early allograft function on renal allograft survival. Transplantation 1997; 63:1611-5.
Fellstrom B. Nonimmune risk factors for chronic renal allograft disfunction [Review]. Transplantation 2001; 71(11 Suppl): SS10-6.
Paul LC. Immunologic risk factors for chronic renal allograft dysfunction. Transplantation 2001; 71(11 Suppl): SS17-23.
Kasiske BL, Ma JZ, Louis TA, Swan SK. Long-term effects of reduced renal mass in humans. Kidney Int 1995; 48: 814-9.
Perez Fontan M. Factores Pronósticos Precoces en el trasplante renal de Cadáver bajo Inmunosupresión con Ciclosporina. Tesis Doctoral. Universidad de La Coruña.Departamento de Ciencias da Saude. Marzo 1997.
Perez Fontan M, Rodríguez –Carmona A, Bouza P, et al. Outcome of grafts with long-lasting delayed function after renal transplantation. Transplantation 1996; 62: 42-7.
Bakir N, Sluiter WJ, Ploeg RJ et al. Primary renal graft thrombosis. Nephrol.Dial & Transplant 1996; 11: 140-7.
Lipkowitz GS, Hong JH, Galishooff ML,et al. Immediate postransplant renal failure: Evidence for an immunologic pathogenesis. Transplant Proc 1989; 21: 1256-57.
Madsen M, Fjeldborg O, Rasmussen A, et al. The impact of immunological risk factors on the outcome of cadaveric renal transplantation as revealed in transplants with primary graft anuria. Transplant Proc 1990; 22: 1371-2.
Marcen R, Orofino L, Orte L. Necrosis tubular aguda en el inmediato postransplante renal. Nefrología 1992; 12 (supl4): 210-15.
Vazquez-Martul E. Patología del trasplante renal. En Trasplante Renal .Gonzalez Martín M. Garcia Buitrón JM, edit., Biblioteca Aula Medica, Madrid, 2000; pp 215-23.
Ojo AO, Wolfe RA, Held PJ, Port FK, Schmouder RL. Delayed graft function: risk factors and implications for renal allograft survival. Transplantation 1997; 63: 968-74.
Ojo AO, Hanson JA, Meier-Kriesche H, Okechukwu CN, Wolfe RA, Leichtman AB, Agodoa LY, Kaplan B, Port FK. Survival in recipients of marginal cadaveric donor kidneys compared with other recipients and wait-listed transplant candidates. J Am Soc Nephrol 2001; 12: 589-97.
Pratschke J, Wilhelm MJ, Kusaka M, Basker M, Cooper DK, Hancock WW, Tilney NL. Brain death and its influence on donor organ quality and outcome after transplantation. Transplantation 1999; 67: 343-8.
Whiting JF, Woodward RS, Zavala EY, Cohen DS, Martin JE, Singer GG, Lowell JA, First MR, Brennan DC, Schnitzler MA. Economic cost of expanded criteria donors in cadaveric renal transplantation: analysis of Medicare payments. Transplantation 2000; 70: 755-60.
Gaber LW, Moore LW, Alloway RR, Amiri MH, Vera SR, Gaber AO. Glomerulosclerosis as a determinant of posttransplant function of older donor renal allografts. Transplantation 1995; 60: 334-9.
Randhawa PS, Minervini MI, Lombardero M, Duquesnoy R, Fung J, Shapiro R, Jordan M, Vivas C, Scantlebury V, Demetris A. Biopsy of marginal donor kidneys: correlation of histologic findings with graft dysfunction. Transplantation 2000; 69: 1352-7.
Bellamy CO, Randhawa PS. Arteriolitis in renal transplant biopsies is associated with poor graft outcome. Histopathology 2000; 36: 488-92.
Mihatsch MJ, Kyo M, Morozumi K, Yamaguchi Y, Nickeleit V, Ryffel B. The side-effects of ciclosporine-A and tacrolimus. Clin Nephrol 1998; 49: 356-63.
Solez K, Racusen LC, Marcussen N, Slatnik I, Keown P, Burdick JF, Olsen S. Morphology of ischemic acute renal failure, normal function, and cyclosporine toxicity in cyclosporine-treated renal allograft recipients. Kidney Int 1993; 43: 1058-67.
Kliem V, Brunkhorst R. Tacrolimus in Kidney Transplantation. A Clinical review. Nephrology Grand Rounds. Clinical Issue. En Nephrology. Ritz E., and Zeier M. editores. Nephron 1998; 79: 8-20.
Iwaki Y, Terasaki PI. Primary nonfunction in human cadaver kidney transplantation: Evidence for hidden hyperacute rejection. Clin Transplantation 1987; 1: 125-31.
Sumrani NB, Delaney V, Daskalakis P, Hong JH et al. The detrimental effect of early rejection on long term renal allograft outcome. Transplant Proc 1992; 24: 1750-52.
Leggat JE Jr, Ojo AO, Leichtman AB, Port FK, Wolfe RA, Turenne MN, Held PJ. Long-term renal allograft survival: prognostic implication of the timing of acute rejection episodes. Transplantation 1997; 63: 1268-72.
Soderberg-Naucler C, Emery VC. Viral infections and their impact on chronic renal allograft dysfunction. Transplantation 2001; 71(11 Suppl): SS24-30.
Cameron J, Rigby RJ, van Deth AG, Petrie JJ. Severe tubulo-interstitial disease in a renal allograft due to cytomegalovirus infection. Clin Nephrol 1982; 18: 321-5.
Randhawa PS, Finkelstein S, Scantlebury V, Shapiro R, Vivas C, Jordan M, Picken MM, Demetris AJ. Human polyoma virus-associated interstitial nephritis in the allograft kidney. Transplantation 1999; 67: 103-9.
Ito M, Hirabayashi N, Uno Y, Nakayama A, Asai J. Necrotizing tubulointerstitial nephritis associated with adenovirus infection. Hum Pathol. 1991; 22: 1225-31.
Randhawa P, Demetris AJ, Pietrzak B, Nalesnik M. Histopathology of renal posttransplant lymphoproliferation: comparison with rejection using the Banff schema. Am J Kidney Dis 1996; 28: 578-84.
Alfrey EJ, Friedman AL, Grossman RA, Perloff LJ, at al. A recent decrease in the time to development of monomorfous and polymorfous posttransplant lymphoproliferative disorder. Transplantation 1992; 54: 250-3.
Kreis HA, Ponticelli C. Causes of late renal allograft loss: chronic allograft dysfunction, death, and other factors. Transplantation 2001; 71(11 Suppl): SS5-9.
Kasiske BL, Kalil RSN, Soon lee H, and Rao KV. Histopathologic findings associated with a chronic, progressive decline in renal allograft function. Kidney International 1991; 40: 514-24.
Furness PN. Histopathology of chronic renal allograft dysfunction. Transplantation 2001; 71(11 Suppl): SS31-6.
Morris RE. Vascular and cellular mechanisms of chronic renal allograft dysfunction. Transplantation 2001, 71: SS37-41.
Castelao AM, Griñó JM, Serón D, Andrés E, et al. Pathological differential diagnosics of proteinuria and late failure after renal transplantation. Transplantation Proc 1992, 24: 110-12.
Halloran PF, Schlaut J, Solez K, Srinivasa NS. The significance of the anti-class I response. II. Clinical and pathologic features of renal transplants with anti-class I-like antibody. Transplantation 1992; 53: 550-5.
Lobo PI, Spencer CE, Stevenson WC, Pruett TL. Evidence demonstrating poor kidney graft survival when acute rejections are associated with IgG donor-specific lymphocytotoxin. Transplantation. 1995; 59: 357-60.
Trpkov K, Campbell P, Pazderka F, Cockfield S, Solez K, Halloran PF. Pathologic features of acute renal allograft rejection associated with donor-specific antibody, Analysis using the Banff grading schema. Transplantation 1996; 61: 1586-92.
Perez Fontan M, Rodriguez-Carmona A, Bouza P, Valdes F. The prognostic significance of acute renal failure after renal transplantation in patients treated with cyclosporin. QJM 1998; 91: 27-40.
Goes N, Urmson J, Ramassar V, Halloran PF. Ischemic acute tubular necrosis induces an extensive local cytokines response. Transplantation 1995; 59: 565-72.
Vazquez-Martul E. Principales patrones histológicos en el Injerto Renal Inicialmente No Funcionante. Comunicación y Abstracts. XVII Congreso Nacional de la Sociedad Española de Anatomia patológica, Barcelona 30 Abril, 1995.
Vazquez-Martul E, Alvarez GA, Lopez M, Rodríguez CA. Principales parámetros histológicos-estructurales de la necrosis Tubular Aguda. Modelo experimental isquémico. Nefrología 1992; 12: 20-9.
Vazquez Martul E. Alteraciones morfológicas del fracaso renal agudo. En: Liaño F, Pascual J, editores. Barcelona: Masson, 2000; p. 59-67.
Olsen S, Burdick JF, Keown PA, Wallace AC, et al. Primary acute renal failure («acute tubular necrosis») in the transplanted kidney: Morphology and pathogenesis. Medicine 1989; 68: 173-87.
Saad R, Gritsch HA, Shapiro R, Jordan M, Vivas C, Scantlebury V, Demetris AJ, Randhawa PS. Clinical significance of renal allograft biopsies with «borderline changes», as defined in the Banff Schema. Transplantation 1997; 64: 992-5.
Colvin RB, Dvorak HF. Basophils and mast cells in renal allograft rejection. Lancet 1974 1; 1: 212.
David-Neto E, Ribeiro DS, Ianhez LE, Palomino S, Saldanha LB, Arap S, Sabbaga E. Acute interstitial nephritis of plasma cells: a new cause for renal allograft loss. Transplant Proc 1993; 25(1 Pt 2): 897-9.
Meehan SM, Domer P, Josephson M, Donoghue M, Sadhu A, Ho LT, Aronson AJ, Thistlethwaite JR, Haas M. The clinical and pathologic implications of plasmacytic infiltrates in percutaneous renal allograft biopsies. Hum Pathol 2001 Feb; 32(2): 205-15.
Colvin RB, Cohen AH, Saiontz C, Bonsib S, Buick M, Burke B, Carter S, Cavallo T, Haas M, Lindblad A, Manivel JC, Nast CC, Salomon D, Weaver C, Weiss M. Evaluation of pathologic criteria for acute renal allograft rejection: reproducibility, sensitivity, and clinical correlation. J Am Soc Nephrol 1997; 8: 1930-41.
van Saase JL, van der Woude FJ, Thorogood J, Hollander AA, van Es LA, Weening JJ, van Bockel JH, Bruijn JA. The relation between acute vascular and interstitial renal allograft rejection and subsequent chronic rejection. Transplantation. 1995; 59: 1280-5.
Schweitzer EJ, Drachenberg CB, Anderson L, Papadimetriou JC, Kuo PC, Johnson LB, Klassen DK, Hoehn-Saric E, Weir MR, Bartlett ST. Significance of the Banff borderline biopsy. Am J Kidney Dis 1996; 28: 585-8.
Colvin RB, Mauiyyedi S. Differential diagnosis between infection and rejection in renal allografts. Transplant Proc. 2001; 33: 1778-9.
Drachenberg CB, Beskow CO, Cangro CB, Bourquin PM, Simsir A, Fink J, Weir MR, Klassen DK, Bartlett ST, Papadimitriou JC. Human polyoma virus in renal allograft biopsies: morphological findings and correlation with urine cytology. Hum Pathol 1999; 30: 970-7.
Tuazon TV, Schneeberger EE, Bhan AK, McCluskey RT, Cosimi AB, Schooley RT, Rubin RH, Colvin RB. Mononuclear cells in acute allograft glomerulopathy. Am J Pathol 1987; 129: 119-32.
Hiki Y, Leong AS, Mathew TH, Seymour AE, Pascoe V, Woodroffe AJ. Typing of intraglomerular mononuclear cells associated with transplant glomerular rejection. Clin Nephrol 1986; 26: 244-9.
Richardson WP, Colvin RB, Cheeseman SH, Tolkoff-Rubin NE, Herrin JT, Cosimi AB, Collins AB, Hirsch MS, McCluskey RT, Russell PS, Rubin RH. Glomerulopathy associated with cytomegalovirus viremia in renal allografts. N Engl J Med 1981; 305: 57-63.
Axelsen RA, Seymour AE, Mathew TH, Canny A, Pascoe V. Glomerular transplant rejection: a distinctive pattern of early graft damage. Clin Nephrol 1985; 23: 1-11.
Maryniak RK, First MR, Weiss MA. Transplant glomerulopathy: evolution of morphologically distinct changes. Kidney Int 1985; 27: 799-806.
Habib R, Antignac C, Hinglais N, Gagnadoux MF, Broyer M. Glomerular lesions in the transplanted kidney in children. Am J Kidney Dis 1987; 10: 198-207.
Crespo M, Pascual M, Tolkoff-Rubin N, Mauiyyedi S, Collins AB, Fitzpatrick D, Farrell ML, Williams WW, Delmonico FL, Cosimi AB, Colvin RB, Saidman SL. Acute humoral rejection in renal allograft recipients: I. Incidence, serology and clinical characteristics. Transplantation 2001; 71: 652-8.
Mauiyyedi S, Pelle PD, Saidman S, Collins AB, Pascual M, Tolkoff-Rubin NE, Williams WW, Cosimi AA, Schneeberger EE, Colvin RB. Chronic humoral rejection: identification of antibody-mediated chronic renal allograft rejection by C4d deposits in peritubular capillaries. J Am Soc Nephrol 2001; 12: 574-82.
Baid S, Pascual M, Cosimi AB, Chung RT, Colvin RB, Tolkoff-Rubin N. Viruses and thrombotic microangiopathy. Transplantation 1999; 68: 710-1.
Baid S, Pascual M, Williams WW Jr, Tolkoff-Rubin N, Johnson SM, Collins B, Chung RT, Delmonico FL, Cosimi AB, Colvin RB. Renal thrombotic microangiopathy associated with anticardiolipin antibodies in hepatitis C-positive renal allograft recipients. J Am Soc Nephrol 1999; 10: 146-53.
Waiser J, Budde K, Rudolph B, Ortner MA, Neumayer HH. De novo hemolytic uremic syndrome postrenal transplant after cytomegalovirus infection. Am J Kidney Dis 1999; 34: 556-9.
Abramowicz D, Prdier O, Marchant A, Florkin S, et al. Induction of thromboses within renal graft by high-dose prophylactic OKT3. Lancet 1992; 339: 777-8.
![]()