
Vol. 35, n.º 4, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 4, 2002 |
M. Gutiérrez Molina
Sección de Neuropatología. Dpto. de Anatomía Patológica. Hospital Universitario La Paz. Madrid.
INTRODUCCION
La Enfermedad con Cuerpos de Poliglucosano del Adulto (ECPA) es una entidad clinicopatológica que asocia variablemente una Enfermedad de Neurona Motora, Demencia, Neuropatía periférica y Disfunción vesical como hechos clínicos mas característicos. Casos aislados han presentado también un síndrome extrapiramidal (70), crisis convulsivas (40) ó atrapamientos nerviosos múltiples (27). Como indica su nombre está definida morfopatológicamente por la presencia de cuerpos de poliglucosano (CP). La revisión de la literatura revela que se han aportado unos 45 casos. El hallazgo de una deficiencia en el enzima ramificante del glucógeno (47,11) así como de diversas mutaciones en el gen del citado enzima (48,91) autoriza a considerar algunos casos como otra forma de expresión de la Glucogenosis tipo IV. Los CP se disponen en las prolongaciones neuronales, rara vez en el soma, a diferencia de los cuerpos amiláceos (CA) que son esencialmente astrocitarios.
Se describe el estudio clinicopatológico de un caso familiar con esta rara enfermedad.
Caso clínico: Mujer de 56 años que consulta por trastornos de la marcha y del lenguaje constatándose también anomalías de funciones cognitivas superiores.
En los antecedentes familiares destaca el fallecimiento de un hermano (60a) con el diagnóstico clínico de ELA-Demencia y el de otra hermana (45a) con posible ELA.
Enfermedad Actual: Desde hace 9 meses presenta dificultad para la marcha, con ampliación de la base de sustentación, lenguaje disártrico y alteraciones de la memoria.
Exploración física sistémica: sin alteraciones significativas.
Exploración neurológica: Facies amímica. Pares craneales normales. Fuerza muscular conservada. Hipertonia. Reflejos osteotendinosos exaltados con aumento del área reflexógena, clonus aquíleo inagotable y Babinski bilateral. Marcha espástica. No trastornos sensitivos ni cerebelosos.
Funciones corticales superiores: Bradipsíquia con alteración en la atención y concentración. Afasia, fluidez verbal escasa y parafasias fonémicas. Apraxias del vestido y de la marcha. Alteración de la conducta alimentaria con deshinibición. No agnosias.
Conclusión del Informe Neuropsicológico: Presencia de signos de disfunción frontal con indicios de daño en lóbulo temporal.
Exploraciones complementarias: Hemograma y bioquímica normales. Hormonas tiroideas, ácido fólico, B12 y serología de lúes normales . Marcadores tumorales negativos. Potenciales evocados visuales, auditivos y somatosensoriales normales. Resonancia Magnética cerebral: Atrofia discreta de predominio frontal.
SPECT: discreta hipocaptación frontal y temporal.
Evolución: En los dos años siguientes, hasta su fallecimiento, se observaron trastornos severos del lenguaje, grave deterioro del razonamiento abstracto y una progresiva dificultad para la marcha, hasta hacer vida cama-sillón, con dependencia completa del cuidador para todas las actividades de la vida diaria. Incontinencia vesical. Progresiva instauración de mutismo, resultando imposible cualquier tipo de intercambio con la paciente. Diez días antes de su último ingreso presentó infección respiratoria. En estado febril y con signos de sepsis, se detectó un importante trastorno hidroelectrolítico, falleciendo la enferma en estado de shock séptico, a los seis días de su ingreso.
Los diagnósticos clínicos con los que se solicitó estudio necrópsico parcial del encéfalo fueron: Probable Demencia Frontotemporal y Esclerosis Lateral Amiotrófica.
Neuropatología: Peso del encéfalo reducido a 950 g debido a una atrofia cerebral de predominio frontal, apreciándose en el exámen externo un notable adelgazamiento de las circunvoluciones y ampliación de los surcos intergirales. (fig. 1). En el examen externo no destacan alteraciones en cerebelo, substancia negra ni polígono de Willis. Al efectuar los cortes seriados se aprecia un adelgazamiento de la corteza frontal y una moderada dilatación de los ventrículos laterales en sus proyecciones frontales.
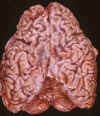
Fig. 1. Atrofia cerebral frontal.
Microscópicamente se observa en la corteza cerebral, sobre todo de la región motora, una perdida neuronal con astrogliosis reaccional. Como hecho marcador diagnóstico se ha considerado la presencia de corpúsculos de poliglucosano que se han observado distribuidos difusamente, con mayor afectación en substancia gris que en substancia blanca. La corteza cerebral, el tálamo, la substancia negra y la capa molecular cerebelosa han sido las estructuras mas afectadas. En la corteza cerebral se encuentran en todo el espesor cortical, libres en el neuropilo (fig. 2) y en las prolongaciones neuronales (fig. 3), aunque también se han observado en el pericarion neuronal comprimiendo y desplazando al núcleo periféricamente (fig. 4). Esta última expresión se ha observado particularmente en la capa molecular del cerebelo.

Fig. 2. Corteza motora cerebral con
cuerpos de poliglucosano. PAS. 50x.

Fig. 3. Cuerpo de poliglucosano
intraaxonal. Antineurofilamentos. 100x.
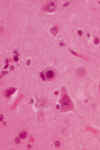
Fig. 4. Cuerpo de poliglucosano en
soma neuronal. HE. 180x.
Otro aspecto destacable ha sido la desmielinización del tracto corticoespinal observada con una intensidad decreciente desde las pirámides bulbares hasta el pié de los pedúnculos cerebrales.
DISCUSIÓN
La Enfermedad con Cuerpos de Poliglucosano del Adulto (ECPA) fué descrita por Suzuki en 1971 (82). Se han aportado unos 45 casos pero únicamente en un tercio de ellos se ha realizado estudio necrópsico (42,56,71,80,54,6,29,51,21, 81,8,7). El hallazgo de CP en el estudio neuropatológico tiene una notable significación diagnóstica.
Los poliglucosanos son moléculas de gran tamaño formadas por polisacáridos anhidros que por hidrólisis dan lugar a una hexosa, fundamentalmente glucosa. El almidón, la celulosa y el glucógeno son poliglucosanos.
La composición y la morfología de los CP son semejantes a la de los Cuerpos Amiláceos (CA). De estos se ha constatado un incremento en relación con la edad, sobre todo a partir de los 40 años (16) por lo que el primer problema que se plantea en el estudio neuropatológico de la ECPA es valorar el significado que pueda tener el hallazgo de estos cuerpos. Dada la semejanza de su estructura y tamaño (variables) , resulta práctico atender esencialmente a su distribución topográfica y a las caraterísticas de la afectación celular. Los CA se situan subpialmente, especialmente en la profundidad de los surcos de la Insula, de la superficie medial de las circunvoluciones temporales y sobre todo en relación con el hipocampo. En la substancia blanca tienden a disponerse alrededor de los vasos medianos y grandes. Otra disposición particularmente frecuente es la yuxtaventricular, sobre todo en relación con el cuerpo calloso (20). En la ECPA, como en el caso que se aporta, los CP se distribuyen en todo el espesor cortical y no se ha apreciado la selectividad topográfica referida en los CA. Microscópicamente los CA se relacionan, salvo excepciones (20), con las prolongaciones astrocitarias (64) mientras que los CP en la ECPA se disponen en las prolongaciones neuronales. En el caso aportado, su localización en el soma neuronal no ha sido tan excepcional como se señala en la literatura. Sobre todo en la capa molecular de la corteza cerebelosa, presumiblemente afectando a las neuronas estrelladas, con frecuencia adoptaban esta disposición, rechazando al núcleo periféricamente (fig. 5). La apariencia es como la que describe Lafora afectando a las «celulas nerviosas pequeñas de la capa granular de la corteza», que puede contemplarse en la lámina II del trabajo que aportó en 1913 titulado »Nuevas investigaciones sobre los cuerpos amiláceos del interior de las células nerviosas»(45).
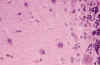
Fig. 5. Molecular cerebelosa con
varias neuronas englobando cuerpos de poliglucosano. HE. 160x.
1. LOS CUERPOS DE POLIGLUCOSANO EN NEUROPATOLOGÍA (tabla I)
Existen distintas enfermedades que están marcadas en el exámen neuropatológico por los CP. Estos pueden diferir en su expresión morfológica, distribución topográfica, tipo celular afecto, disposición intraneuronal y contexto clínico en el que se desarrollan.

En la Epilepsia Mioclónica de Lafora (44), el hallazgo en el soma neuronal de uno ó de varios cuerpos de poliglucosano constituye el marcador diagnóstico neuropatológico (cuerpos de Lafora). La corteza cerebral, el tálamo, la substancia negra y el núcleo dentado son las estructuras mas afectadas por el depósito (fig. 6).
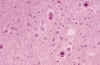
Fig. 6. N. Dentado en Epilepsia mioclónica de Lafora con variada
expresión morfológica de los corpúsculos. HE. 100x.
Los cuerpos de Bielschowsky también son intraneuronales y se observan en el Pallidum externo de pacientes con Coreo-Atetosis, algunos de los cuales presentan Status Marmoratus en los núcleos basales. (3,88).
En la Amilopectinosis ó Glucogenosis tipo IV la afectación del SNC se ha descrito en muy pocos casos. El depósito es difuso, con características análogas a la distribución de los de CA «fisiológicos». Se observa afectación de los ganglios basales y de la corteza cerebral, tanto de sus capas profundas como de las superficiales. (73,76). Se consideraba que el tipo celular afectado era el astrocito (52), sin embargo Gallen describió afectación neuronal en una forma muy severa de Amilopectinosis (25). Mas recientemente se ha descrito un caso neonatal con extenso acúmulo neuronal de poliglucosanos en una forma letal de Glucogenosis mixta, con deficiencia de Enzima Ramificante y de Fosforilasa. (35).
La observación en un paciente con Mixedema y Ataxia de «neural myxedema bodies» semejantes a los CA ha resultado anecdótica (59).
Entre las Neurodegeneraciones se ha descrito la existencia masiva de CA en un paciente con Atrofia Pallido-Nigro-Luysiana (41) y en un caso de Atrofia OlivoPontoCerebelosa en la capa molecular del cerebelo (57).
Aunque muy raras, existen aportaciones sobre Esclerosis Lateral Amiotrófica con cuerpos mioclónicos de Lafora que considero de difícil tipificación. (5). Venia refiriéndose en sucesivas ediciones del Greenfield’s Neuropathology (3.ª, 4.ª y 5.ª) que los CA son particularmente numerosos en procesos degenerativos tales como la Enfermedad de Neurona Motora. Sin embargo, esta observación no ha sido recogida en su 6ª edición (42). Tampoco en descripciones clásicas de la ELA se considera su presencia de forma significativa (36,38) y un estudio reciente ha descartado que exista un incremento de CA en la ELA. (19).
2. LOS CUERPOS AMILÁCEOS Y LA ESCUELA DE CAJAL
a. Sobre su composición: Amiloide vs Amiláceo
Giambattista Morgagni describió en 1723 los cuerpos amiláceos de la próstata y en 1837 Purkinje descubrió los CA de los centros encefálicos (68). En 1854 Virchow consideró derivadas de la celulosa tanto las inclusiones amiláceas del sistema nervioso como una substancia hialina a la que denominó Amiloide. (68). La atribuida semejanza con el almidón, otro polímero de la glucosa constituido por Amilosa y por Amilopectina, podía justificar sus nombres pero creó una prolongada confusión que afectó al propio Cajal quién refiriéndose a ellas afirmaba: «La amilosis ó degeneración amiloidea consiste en el depósito en torno a los vasos sanguíneos de una materia proteica homogénea y transparente, que se aproxima al almidón por su propiedad de teñirse en rojo caoba por el iodo ... En estado normal contienen la materia amiloide la próstata y los centros nerviosos...Semejantes a las prostáticas son las concreciones amiloides del cerebro y médula, cuya residencia mas común es la vecindad del epéndimo y los territorios de la substancia gris próximos a las meninges.» Por la descripción efectuada resulta evidente que Cajal consideraba la substancia amiloide y los cuerpos amiláceos como un mismo substrato químico, tal y como confirma el párrafo siguiente: «El nombre que lleva la substancia amiloide ó amilácea, le fue dado por Virchow, por la singular propiedad que posee de teñirse en rojo caoba por el iodo a semejanza del almidón».
Sin embargo Cajal objetaba y enseñaba que: «en realidad la reacción no es idéntica, pués como es sabido , el almidón reacciona con el yodo ya en violado azul, ya en azul puro; pero la semejanza aumenta si...se trata la materia amiloidea por el ácido sulfúrico». Al observar que «los cuerpos estratificados amiláceos reaccionan raramente en azul ó violeta, aún después de la acción del ácido sulfúrico¨ concluye Cajal que «la materia amilácea no puede estimarse, como pensaba Virchow, como un principio vecino de la celulosa, sino como un principio especial albuminoide»(61).
¿Interpretación equivocada de los hechos por parte de Cajal ?
En 1913 todavía no estaba definida la separación de Amiloide y Amiláceo como puede deducirse por el comentario de Lafora sobre el material que se acumula en la epilepsia mioclónica que lleva su nombre: «...para evitar confusiones conviene llamar Amilácea a esta substancia nitrogenada y Amiloide a la hidrocarbonada que se asemeja al almidón y que es la que constituye la degeneración amiloide» (45).
Actualmente se considera que los Cuerpos Amiláceos (CA) son esencialmente Polisacaridos que al estar constituidos por polímeros de glucosa se llaman también Poliglucosanos. La interpretación de Virchow sobre la Amiloide era errónea pero su opinión acerca de la naturaleza de los CA era mas acertada. Sin embargo, hoy se sabe que los CA tienen una fracción proteica que se estima es de un 4,7% (72) y se ha verificado inmunohistoquímicamente que además de Ubiquitina, Tau y proteina heat shock, contienen también un componente P amiloide (78). ¿Era lo que Cajal detectaba distrayéndole en el análisis de la composición global de los CA?
b. Sobre el origen de los CA
Se han contemplado diversas teorías con respecto al origen de los cuerpos amiláceos. La especial topografía que tienen los CA «fisiológicos», relacionados con el envejecimiento, soportó la idea de un origen glial. Según Del Río , el mas decidido defensor de esta teoría fué Obersteiner , para quién «los CA se presentarían en número variable dentro de las células neurogliales y cuando estas se destruyen quedarían libres» (68).
Robertson en 1900 propuso un origen de los CA en un nuevo tipo de celula, muy someramente descrita por él mismo, «en una sola página» según Del Río, (69), a la que denominó Mesoglia, de supuesta estirpe mesodérmica. Dos décadas después, las profundas investigaciones de Pio del Rio-Hortega concernientes a la microglía (66,67) , fundamentadas en los trabajos previos de Achúcarro sobre las «celulas en bastoncito» (1,2), fueron consideradas por Cajal como referidas a la Mesoglía de Robertson (63). Esta aportación de Cajal hirió el carácter susceptible de Pío del Río quién consideró que «el primordial objeto era despojarme de la prioridad del descubrimiento de la Microglía y atribuírsela a Robertson» (69). Convencido de la naturaleza glial de este tipo celular así como de su correspondencia con la glía de escasas radiaciones ú oligodendroglía, era la Microglía por él investigada la que debía quedar como el verdadero elemento de origen mesodérmico. El reconocimiento de Cajal, en carta personal dirigida a Del Rio, a los pocos dias, sin mención alguna a su «Tercer elemento del sistema nervioso», restañó la herida sensibilidad del discípulo y puso de manifiesto el espiritu justo y generoso del maestro.
También Gonzalo R Lafora , en un estudio sobre CA realizado en 1911, describió la existencia de «células neuróglicas cuyo citoplasma se halla cargado de pequeños cuerpos amiláceos» (43) . Fue el mismo año de su importante trabajo sobre la Epilepsia Mioclónica, que he podido consultar en la biblioteca del Instituto Cajal. En el pie de la primera página se puede leer «Al Prof Don Santiago R y Cajal. Homenaje del autor» (44). Desconozco si Lafora pudo enseñarle a Cajal las preparaciones histológicas que fundamentaron su histórica aportación, elaborada siendo histopatólogo del «Governement Hospital for the Insane» en Washington.
Tras las publicaciones fundamentales sobre la Microglía Del Río aportó un trabajo titulado «Papel de la microglía en la formación de los cuerpos amiláceos del tejido nervioso». (68). Sobre la identificación del tipo celular involucrado, decía : «Las investigaciones efectuadas por nosotros en diferentes casos...nos permitieron observar...cuerpos amiláceos junto a núcleos pequeños y obscuros, frecuentemente aplanados, diferentes a los de la neuroglia común, presentando aspectos muy semejantes a algunos copiados por Lafora» ... «la comprobación de que ...correspondían a elementos microgliales solamente hemos podido efectuarla...con nuestra técnica selectiva para la microglía». Sobre los efectos de los CA, comentaba: «Cuando por crecimiento de las concreciones amiloideas, el núcleo es comprimido demasiado...llega a desaparecer» ...A propósito de la supuesta naturaleza de los CA consideraba «mas lógico que sean productos de desintegración nerviosa los materiales de que se forman las concreciones amiláceas» y sobre el proceso propiamente dicho de constitución de los CA para Del Río «parece probable que se trate de una impregnación del protoplasma... por productos disueltos que adquieren después la forma globulosa» ...aceptando en todo o en parte las ideas de Alzheimer. Comentaba Del Río lamentandose «nosotros que jamás hemos tenido la fortuna de observar cuerpos amiláceos intraaxónicos ni intraneuronales no podemos dudar de su existencia después de haber visto las ilustraciones que figuran en las publicaciones de Lafora». En 1925 ya hacía cinco años que Del Río había pasado a dirigir el laboratorio de Histopatolgía en la «Colina de los Chopos», muy cerca del Hipódromo. Estaba instalado en el «Trasatlántico» de la Residencia de Estudiantes por decisión de Cajal que era el presidente de la Junta de Ampliación de Estudios, de la que dependía. Puede ser que en 1925 Gonzalo R Lafora ya no fuera el responsable del laboratorio de Fisiología y Anatomía de los centros nerviosos, en la misma Residencia, pero algunos años si que debieron coincidir. ¿Por qué razón no vió Del Río las preparaciones de la epilepsia mioclónica? Debe reconocerse que también la lámina que ilustra diversos aspectos de la microglía provista de inclusiones amiláceas en el trabajo citado de Del Río resulta muy convincente. Sin embargo, en el caso que se aporta la microglía no parece tener un papel significativo en relación con los poliglucosanos (fig. 7).
Fig. 7. Molecular cerebelosa con
cuerpo de poliglucosano sin relación aparente con células microgliales. Anti
LCA. 160x.
Quizás en esta época tuviese ya olvidado Del Río algún comentario de Nicolás Achúca rro, con quién comenzó su labor investigadora, acerca de la génesis de los cuerpos amiláceos ya que fue éste uno de los primeros autores en constatar su posible situación intraaxonal (18).
Trabajó Achúcarro en varios laboratorios y «singularmente en el del profesor Alzheimer de quíen fue discípulo predilecto» (62). Posteriormente se incorpora a los dos laboratorios de Cajal, el de investigaciones biológicas y de la catedra, en ambos sin remuneración (55). Logró Cajal «obligarle a regentar una plaza de Auxiliar numerario en espera de la creación de la cátedra de Neuropatología» (62). A Achúcarro debemos que el propio Cajal se sintiese neuropatólogo tal y como leemos en su carta de adhesión al banquete celebrado en honor a Río-Hortega en 1932 «Y el acto será presidido en espíritu por el noble y malogrado Achúcarro, a cuyas iniciativas y consejos tanto debemos los neuropatólogos españoles».
Por iniciativa de Cajal, el equipo de colaboradores constituido alrededor de Achúcarro, se incorporó al grupo del Laboratorio de Investigaciones Biológicas, en el Museo Velasco, pero tras su prematura muerte, ocurrida el 23 de abril de 1918, empezó a sopesar Cajal la conveniencia de una separación efectiva de ambos grupos. Esta tuvo lugar en octubre de 1920. La causa inmediata fueron unos supuestos juicios emitidos por Del Río «extremadamente desdeñosos» acerca de su persona , según le expresaba Cajal en carta fechada, parece ser, el 9 de octubre. A pesar de todo acababa diciéndole que iba a gestionar «de la Junta la adquisición de un local donde pueda Vd...». En su respuesta Del Río afirmaba «rotundamente que cuanto de mí le han referido es completamente falso» terminando la carta «su siempre devoto y agradecido discípulo», con fecha 5 de octubre de 1920.
Desconciertan las fechas correspondientes a ambas cartas recogidas en el ensayo autobiográfico inconcluso de Pio Del Río-Hortega «El Maestro y yo». (69).
3. ECPA: SOBRE LA ASOCIACIÓN ENM-DEMENCIA (tabla II)
En la ECPA resulta frecuente la asociación de una Enfermedad de Neurona Motora a un deterioro cognitivo, generalmente expresado como una demencia ligera o moderada. Ocasionalmente se ha referido como una demencia frontal. (9,70). El hallazgo de los CP constituye una importante ayuda en el diagnóstico neuropatológico ya que la asociación de ENM y demencia plantea un importante problema diagnóstico diferencial. El análisis de las alteraciones cognitivas en la ELA revela que en un 3%-6% de los casos esporádicos coexiste una demencia, alcanzando hasta un 15% en las formas familiares (58). Generalmente se trata de una demencia frontotemporal. Los aspectos neuropatológicos son variados. La presencia de celulas balonizadas y de cuerpos de Pick, ubiquitina y Tau positivos, perfila una patología del tipo de la clásica enfermedad de Pick. En otros pacientes, la observación de inclusiones citoplasmáticas ubiquitinadas en las neuronas corticales permite considerar una demencia tipo ENM, ya que son como las inclusiones característicamente observadas en las motoneuronas espinales de la ELA. En otros casos la neuropatología carece de hechos específicos apreciándose como substrato de la demencia, una perdida neuronal y astrogliosis.

En la ELA-Demencia de Guam y en el Complejo ELA-Parkinson-Demencia se constata como un hecho característico una degeneración neurofibrilar difusa.
En la enfermedad de Alzheimer se describe un incremento numérico de CA (22) pero el conjunto neuropatológico elimina cualquier duda diagnóstica. Tambien en la Demencia vascular pueden observarse numerosos CA, pero topográficamente están relacionados con la patología isquémica.
4. LA ECPA COMO ENFERMEDAD SISTÉMICA
a. Indicación de biopsias diagnósticas
Solamente en un tercio de los 45 casos aportados se ha efectuado estudio necrópsico lo que ha permitido constatar que también existe afectación de nervio periférico, músculo esquelético, miocardio e hígado en esta enfermedad. En la mayoría de los casos el diagnóstico se ha establecido por biopsia del nervio sural. Sin embargo, recientemente se ha efectuado estudio complementario con biopsia cutánea de región axilar cuyos resultados permiten aconsejar su indicación prioritaria. Se pueden observar abundantes cuerpos de inclusión en las celulas mioepiteliales de las glandulas apocrinas, solo unos pocos y mas pequeños en las de las ecrinas y no se encuentran en los conductos ecrinos (15,53), hecho éste último que sí sucede en la enfermedad de Lafora (12,31), cuya afectación glandular apocrina (fig. 8) se considera comparable (15) Para evitar confundir los cuerpos de inclusión, con el material de secreción, es recomendable atender a su habitual relación con la célula mioepitelial y a su mucha mayor positividad con la técnica de PAS (15,53). Si es necesario puede recurrirse a la técnica de Best que también tiñe intensamente las inclusiones.

Fig. 8. Observaciòn con
microscopia electrónica de cuerpo de Lafora relacionado con la célula
mioepitelial de acino glandular cutáneo. 5200x.
El hallazgo de algún CP axonal en el nervio sural o en los fascículos nerviosos observados en las biopsias musculares, no es especifico. Pueden apreciarse en personas sanas (24) o con diabetes (49) y también en otros procesos (29). Se recomienda fundamentar el diagnóstico en la observación de mas de un CP por fascículo, en su detección fuera del axón o en la apreciación de CP de tamaño superior a 30 micras (8,53,70). En la forma juvenil de la Amilopectinosis, causada por una deficiencia completa del enzima ramificante, los CP se observan en axones y células de Schwann pero sobre todo en el perineuro y en las células musculares lisas de los vasos epineurales (73).
Aunque para establecer el diagnóstico de ECPA deban ser las biopsias de piel y de nervio las que se indiquen con preferencia es conveniente saber que en esta enfermedad también otros órganos, sobre todo hígado, músculo esquelético y miocardio, pueden acumular poliglucosanos (8,54).
b. La patología muscular en la ECPA (tabla III)
La expresión del acúmulo de poliglucosanos en el músculo de los pacientes con ECPA es semejante a la descrita en la miopatia con cuerpos de poliglucosano del adulto. Esta, puede ser la expresión de una variante de Amilopectinosis (10,73) pero también se ha descrito en pacientes con una actividad normal de enzima ramificante (28,87). Debe destacarse que la mayoría de las aportaciones sobre este último proceso se han efectuado con una terminología diversa, como: Degeneración basófila muscular (23), Degeneración basófila del músculo esquelético en miopatia hipotiroidea (37), Degeneración basófila en miopatia cardioesquelética (33) (fig. 9), Miopatia con acúmulo de polisacaridos (83) y Polisacaridosis con miopatia cardioesquelética (30). Aunque de forma excepcional, también la patología muscular observada en la enfermedad de Lafora puede ser similar (32). En estas aportaciones generalmente no consta el estado de la actividad enzimática ramificante. Aunque la mayoría de los casos con degeneración basófila asocian un hipotiroidismo, su significado en el contexto de la miopatia hipotiroidea es poco frecuente. La patología habitual de esta miopatia se expresa en las fibras musculares con acúmulos subsarcolémicos de glucogeno. En los casos con degeneración basófila (léase con acúmulo de poliglucosano) debe añadirse algún otro factor que interfiera la actividad del enzima ramificante y que futuras investigaciones podrán dilucidar.

Fig. 9. Cuerpos poliglucosano en
Degeneración basófila muscular en paciente con Cardiomiopatía esquelética.
HE 160x.
5. ASPECTOS ETIOPATOGÉNICOS DE LA EPCA
a. Casos definidos de Amilopectinosis
Los hallazgos enzimáticos y genómicos obtenidos en algunos casos de ECPA, aportados en la última década (47,11,48,91) permiten considerarlos como una forma de Glucogenósis tipo IV. En 1991 se constató, por primera vez, una disfunción hereditaria del enzima ramificante que podría estar implicada en la etiopatogenia de esta enfermedad. Se trataba de dos pacientes no relacionados entre sí, de origen judio Ashkenazi y de padres consanguíneos, cuya actividad leucocitaria enzimática era de un 15%, mientras que la de sus hijos era de un 50-60%, con respecto a controles (47). Los inmediatos resultados posteriores confirmaron este hallazgo pero únicamente en pacientes con esta procedencia étnica (11). En 1998 se considera por primera vez la ECPA como una variante alélica de la Glucogenósis Tipo IV al detectarse (48), en el gen que codifica el enzima ramificante la misma mutación Tyr 329 Ser que se había identificado dos años antes en una forma hepática no progresiva de esta glucogenosis. (4,50). Recientemente se ha descrito por primera vez en un paciente caucásico la misma deficiencia enzimática con dos mutaciones distintas (Arg515His y Arg524Gln) en el gen del enzima ramificante (91). Pero ¿cual es la etiopatogenia del proceso en aquellos casos en los que no se han detectado estas anomalías?
b. Teoría del desequilibrio enzimático Glucógenosintetasa-Enzima Ramificante
Esta teoría sería aplicable a aquellos casos que tienen una actividad enzimática ramificante normal.
En la Patología de los errores congénitos del metabolismo la expresión morfopatológica del depósito de una substancia puede ser semejante aunque las causas sean diversas. Por ejemplo, es muy significativa la notable variabilidad genómica de las Ceroidelipofuscinosis que, sin embargo, están caracterizadas por el depósito de un material con análogas características histoquímicas.
Con respecto a los poliglucosanos hace ya mas de veinte años que su depósito, en una miopatia del adulto causada por la deficiencia de fosfofructoquinasa (34) fue atribuida a un desequilibrio entre las actividades enzimáticas de la glucogenosintetasa y del enzima ramificante, inducido por unos niveles intramusculares elevados de glucosa 6 fosfato. En este mismo sentido apuntan estudios efectuados en un modelo de ratón knock-out con enfermedad de Pompe, cuyos resultados aportan «sorpresas de la ingeniería genética» consistentes en que la sobreexpresión provocada de glucogenosintetasa causa un acúmulo en las fibras musculares indistinguible de los CP (60). Los autores proponen la misma hipótesis del desequilibrio entre la glucogenosintetasa y la actividad del enzima ramificante, no solo como mecanismo en la producción de estos cuerpos en los ratones sino también en algunos de los desordenes clínicos en los que el hallazgo de poliglucosanos es un hecho frecuente u obligado, entre los que incluyen la enfermedad de Lafora, la Glucogenosis tipo IV, la ECPA y la Glucogenosis tipo VII ó deficiencia de fosfofructoquinasa.
c. Contribución de la enfermedad de Lafora al conocimiento de la EPCA
Ha habido quien ha considerado la ECPA como una forma de Enfermedad de Lafora (80). Sin embargo los pocos casos de inicio tardío que se han descrito de esta enfermedad presentan sustancialmente la misma expresión clínica (epilepsia mioclónica) mientras que en la ECPA son excepcionales las crisis epilépticas.
En la enfermedad de Lafora, a pesar de la composición esencialmente amilopectínica de los cuerpos de inclusión, los resultados de las investigaciones llevadas a cabo sobre su hipotética relación con la Amilopectinosis, han sido siempre negativos.
Desde hace varias décadas se vienen aportando publicaciones que hacen referencia a esta eventualidad. La semejanza histoquímica, bioquímica, inmunohistoquímica y estructural, tanto con microscopia óptica como electrónica, entre los CA, los cuerpos de Lafora y los depósitos de la Amilopectinosis se ha citado repetidamente (72,90).
La localización del gen EPM2A (locus 6q23-25) responsable de la enfermedad de Lafora fue aportada en 1995 por Serratosa (74). El hallazgo subsiguiente por este mismo autor de una microdeleción en la región crítica de este gen que codifica una proteina tirosina fosfatasa (Laforina), sugería que la enfermedad de Lafora pudiera deberse a una inactivación de esta proteina, que debia tener una función importante en el metabolismo del glucogeno (75). Actualmente hay descritas varias microdeleciones y mutaciones puntuales que pueden afectar la funcionalidad de la Laforina. Se trata de una proteína de 331 aminoácidos que en su terminación carboxílica tiene actividad fosfatasa y por su extremo aminoterminal puede unirse al glucógeno. Se localiza en los compartimentos subcelulares específicos en los que la glucogenosintetasa efectua la síntesis del glucógeno. (86).
La Laforina forma parte de una subfamilia que posee la doble capacidad de actuar sobre residuos de tirosina y de serina-treonina (26).
d. Posibles errores causales del acúmulo de poliglucosanos, en base a la teoria del desequilibrio enzimático
d.1. Elementales bases metabólicas sobre la síntesis del glucógeno
En la síntesis del glucógeno desempeña un papel esencial la glucogenosintetasa que en su forma activa se encuentra defosforilada mientras que la forma inactiva está fosforilada. En estos procesos intervienen la proteina fosfatasa I y la glucogenosintetasakinasa 3 (GSK 3) respectivamente.
A su vez, la fosforilación de la GSK 3 en diferentes residuos, de tirosina o de serina-treonina, controla diferencialmente su actividad enzimática. La fosforilación en tirosina provoca su activación mientras que la fosforilación en serina-treonina causa su inactivación.
La inactivación de la GSK 3 determina que la glucogenosintetasa, al no resultar fosforilada, permanezca en su forma activa incrementándose la síntesis de glucogeno.
d.2. Hipótesis etiológicas
Se ha demostrado experimentalmente que una alteración en la actividad de proteina tirosina fosfatasa aumenta la síntesis de glucógeno (46). Se entendería que en la enfermedad de Lafora las distintas anomalías descritas en el gen de la Laforina pudieran tener el efecto referido. En la ECPA podría ser alguna otra proteina tirosina fosfatasa la que estuviese involucrada.
Sin embargo, desconociéndose los mecanismos a través de los cuales la Laforina defectuosa pueda provocar el aumento en la síntesis de glucógeno, podría contemplarse como una posibilidad su incapacidad para activar la GSK 3 que conllevaría el que no se fosforilase la glucogenosintetasa y que continuase ésta en su forma activa.
El error en la ECPA podría radicar primariamente en la propia GSK 3.
d.3. Otras obsevaciones
El significado etipatogénico de la diabetes referida en varios pacientes con ECPA (6,29,71,82,85) constituye otro interesante aspecto a investigar dadas las posibles implicaciones en el conocimiento global de estas patologías.
BIBLIOGRAFÍA
Achucarro N: Sur la formation de cellules á batonnet (stabchenzellen) et d’autres elements similaires dans le systeme nerveux central. Laboratoire d’Investigations Biologiques VI: 7-122, 1908.
Achúcarro N: Algunos datos relativos a la naturaleza de las células en bastoncito de la corteza cerebral humana obtenidos con el método de Cajal. Trab Lab Investig Biol Univ Madr VIII: 169-176, 1910.
Adler D, Horoupian D S, Towfighi J, et al: Status marmoratus and Bielschowsky bodies. A report of two cases and review of the literature. Acta Neuropathol 56: 75-77, 1982.
Bao Y, Kishnani P, Wu J-Y, Chen Y-T: Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest 97: 941-948, 1996.
Barz H, Kemmer CH, Kunze D, Sachs B: Amyotrophe lateralsklerose mit myoklonuskorpern. Zbl Allg Path Bd 120: 333-342, 1976.
Baudrimont M, Gherardi R, Gray F et al: Adult poliglucosan body disease (APBD): Report of 3 cases. (Abstract). J Neuropathol Exp Neurol 45: 336, 1986.
Berkhoff M, Weis J, Schroth G, Sturzenegger M: Extensive white matter changes in case of adult polyglucosan body disease. Neuroradiology 43: 234-236,2001.
Bigio EH, Weiner MF, Bonte FJ, White CL: Familial dementia due to adult polyglucosan body disease. Clinical Neuropathol 16: 227-234, 1997.
Boulan-Predseil P, Vital A, Brochet B et al: Dementia of frontal lobe type due to adult polyglucosan body disease. J Neurol 242: 512-516, 1995.
Bornemann A, Besser R, Shin YS,Goebel HH: A mild adult myopathic variant of type IV glycogenosis. Neuromusc Disord 6: 95-99, 1996.
Bruno C, Servidei S, Shanske S et al: Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol 33: 88-93, 1993.
Busard BLSM, Renier WO, Gabreels FJM et al: Lafora’s disease: comparison of inclusion bodies in skin and in brain. Arch Neurol 43: 296-299, 1986.
Busard HLSM, Gabreels-Festen AAVM, Renier WO et al: Axilla skin biopsy: a reliable test for the diagnosis of Lafora’s disease. Ann Neurol 21: 599-601, 1987.
Busard HLSM, Gabreels-Festen AAWM, van’t Hof MA et al: Polyglucosan bodies in sural nerve biopsies. Acta Neuropathol 80: 554-557, 1990.
Busard HLSM, Gabreels-Festen AAWM, Renier WO et al: Adult poliglucosan body disease: the diagnostic value of axilla skin biopsy. Ann Neurol 29: 448-451, 1991.
Busard HLSM, Span JPT, Renkawek, Renier WO et al: Polyglucosan bodies in brain tissue: a systematic stydy. Clin Neuropathol 13: 60-63, 1994.
Cafferty M S, Lovelace R E, Hays A P et al: Polyglucosan body disease. Muscle Nerve 14: 102-107, 1991.
Catola G, Achúcarro N: Uber die entstchung der amyloidkorperchen im zentralnervensystem. Virchows Archiv f Path Anat 184: 454-469, 1906.
Cavanagh JB: Spinal corpora amylacea and motor neuron disease: a quantitative study. J Neurol Neurosurg Psychiat 65: 488-491,1998.
Cavanagh JB: Corpora amylacea and the family of polyglucosan diseases. Brain Res Rev: 29: 265-295, 1999.
Chretien F, Louarn F, Lescs MC, Gray F: La maladie des corps polysaccharidiques. Arch Anat Cytol Path 41: 9-17, 1993.
Cisse S, Perry G, Lacoste-Royal G et al: Immunochemical identification of ubiquitin and heat-shock proteins in corpora amylacea from normal aged and Alzheimer’s disease brains. Acta Neuropathol 85: 233-240, 1993.
Ewing SL, Rosai J: Basophilic (mucoid) degeneration of skeletal muscle. Arch Pathol 97: 60-62.
Fukuhara N: Intra-axonal corpora amylacea in the peripheral nerve seen in a healthy woman. J Neurol Sci 34: 423-426, 1977.
Gallen CC, Schultz P, Thomas CS, et al: Glycogenosis IV: Clinical and pathological findings in siblings. Ann Neurol 20: 404, 1986.
Ganesh S, Agarwala KL, Ueda K et al: Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum Mol Genet 9: 2251-2261, 2000.
Gil-Neciega E, Pareja JA, Chinchón I, et al: Neuropatía con atrapamientos múltiples en la enfermedad con cuerpos de poliglucosano del adulto. Neurología 10: 167-170, 1995.
Goebel HH, Shin YS, Gullota F et al: Adult polyglucosan body myopathy. J Neuropathol Exp Neurol 51: 24-35, 1992.
Gray F, Gerardi R, Marshall A et al: Adult Polyglucosan body disease. J Neuropathol Exp Neurol 4 : 459-474, 1988.
Greene GM, Weldon DC, Ferrans VJ et al: Juvenile polysaccharidosis with cardioskeletal myopathy. Arch Pathol Lab Med 111: 977-982,1987.
Gutierrez Molina M, Santamaria Blanco P, Leon Arencibia L et al: Enfermedad de Lafora. Análisis morfopatológico de la hipotética relación con la Amilopectinosis, a propósito del estudio de tres casos. Arch de Neurobiol 49: 251-267, 1986.
Gutierrez Molina M, Martinez P, Jiménez F, Morales C: La patología muscular en la enfermedad de Lafora, otro nexo de unión con la Amilopectinosis. A propósito de un estudio necrópsico. Arch Neurobiol 50: 56-57, 1987.
Gutierrez Molina M, Leon Arencibia L, Alba Losada J et al: Degeneración basófila del músculo esquelético. A propósito de un caso con miopatía cardioesqueletica. Arch de Neurobiol 50: 25-32, 1987.
Hays AP, Hallet M, Delfs J et al: Muscle phosphofructokinase deficiency: abnormal polysaccharide in a case of late-onset myopathy. Neurology 31: 1077-1086,1981.
Herrick MK, Twiss JL, Vladutiu GD et al: Concomitant Branching enzyme and phosphorilase deficiencies. An unusual glycogenosis with extensive neuronal polyglucosan storage. J Neuropathol Exp Neurol 53: 239-246, 1994.
Hirano A, Malamud N, Kurland LT, Zimmerman HM: A review of the pathologic findings in amyotrophic lateral sclerosis. Cap 5 en Motor Neuron Diseases. Norris FH, Kurland LT ed. 1969.
Ho KL: Basophilic degeneration of skeletal muscle in hypothyroid myopathy. Arch Pathol Lab Med 108: 239-248, 1984.
Hudson AJ: Amyotrophic Lateral Sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain 104: 217-247, 1981.
Karpati G, Carpenter S, Wolfe LS, Sherwin A: A peculiar polisaccharide accumulation in muscle in a case of cardioskeletal myopathy. Neurology 19: 553-564, 1969.
Karpati G, Carpenter S: The clinical spectrum of adult polyglucosan body disease.(Abstract). Neurology 33 (Suppl 2): 246, 1983.
Kosaka K, Matsushita M, Oyanagy S et al: Pallido-nigro-luysial atrophy with massive appearance of corpora amylacea in the CNS. Acta Neuropathol 53: 169-172, 1981.
Kreutzberg GW, Blakemore WF, Graeber MB: Cellular pathology of the central nervous system. Cap 3 en Greenfield’s Neuropathology. Graham DI, Lantos PL ed. Arnold 1997.
Lafora GR: Uber das vorkommen amyloider korperchen im inner der ganglienzellen; Ein beitrag zum studium der amyloiden substanz im nervensystem. Virchow’s Archiv CCV: 295-303, 1911.
Lafora GR, Glueck B: Beitrag zur Histopathologie der myoklonischen epilepsie. Zeitschr.f.ges.Neurol.u.Psych. Bd VI: 1-14, 1911.
Lafora GR: Nuevas investigaciones sobre los cuerpos amiláceos del interior de las células nerviosas. Trab Lab Invest Biol. Univ Madr tomo XI: 29-42, 1913.
Li ZG, Quiang X, Sima AA, Grunberger G: C-peptide attenuates protein tyrosine phosphatase activity and enhances glycogen synthesis in L6 myoblasts. Biochem Biophys Res Commun 280: 615-619, 2001.
Lossos A, Barash V, Soffer D et al: Hereditar branching enzyme dysfunction in adult polyglucosan body disease: a possible metabolic cause in two patients. Ann Neurol 30: 655-662, 1991.
Lossos A, Meiner Z, Barash v et al : Adult polyglucosan body disease in Ashkenazy jewish patients carrying the Tyr 329 Ser mutation in the glycogen branching enzyme gene. Ann Neurol 44: 867-872, 1998.
Mancardi G L, Schenone A, Tabaton M et al: Polyglucosan bodies in the sural nerve of a diabetic patient with polyneuropathy. Acta Neuropathol 66: 83-86, 1985.
McConkie-Rosell A, Wilson C, Piccoli DA, et al: Clinical and laboratory findings in four patients with the non progressive hepatic form of type IV glycogen storage disease. J Inherit Metab Dis 19: 51-58, 1996.
McDonald TD, Faust PL, Brubo C et al: Polyglucosan body disease simulating amyotrophic lateral sclerosis. Neurology 43: 785-790, 1993.
McMaster KR, Powers JM, Hennigar GR et al: Nervous system involvement in Type IV glycogenosis. Arch Pathol Lab Med 103: 105-111, 1979.
Milde P, Guccion JG, Kelly J et al :Adult polyglucosan body disease. Diagnosis by sural nerve and skin biopsy. Arch Pathol Lab Med 125: 519-522, 2001.
Okamoto K, Llena JF, Hirano A: A type of adult polyglucosan body disease. Acta Neuropathol 58: 73-77, 1982.
Oliva Aldamiz H: Cajal y la anatomía patológica española, una historia compartida. Salvat editores S.A.1984.
Peress N, Di Mauro S, Roxburgh VA: Adult polysaccharidosis. Clinicopathological, ultrastructural and biochemical features. Arch Neurol 36: 840-845,1979.
Petito CK, Hart MN, Porro RS, Earle K M: Ultrastructural studies of olivopontocerebellar atrophy. J Neuropathol Exp Neurol 32: 503-522, 1973.
Portet F, Touchon J, Camu W: SLA et troubles cognitifs: revue et analyse de la literature. Rev Neurol 157: 139-150, 2001.
Price TR, Netsty MG: Myxedema and ataxia. Neurology 16:957-962, 1966.
Raben N, Danon M, Lu N et al: Surprises of genetic engineering. A possible model of polyglucosan body disease. Neurology 56: 1739-1745, 2001.
Ramón y Cajal S: Amilosis. Cap III en Manual de Anatomia Patológica General. 3.ª ed.1900.
Ramón y Cajal S: Nicolás Achúcarro. Bol Soc Esp Biol 1918. pág 1-6.
Ramón y Cajal S: Algunas consideraciones sobre la mesoglia de Roberston y Rio-Hortega. Trab Lab Investig Biol Univ Madr XVIII: 109-127, 1920.
Ramsey H: Ultrastructure of corpora amylacea. J Neuropathol Exp Neurol 24: 25-39, 1965.
Rifai Z, Klitzke M, Tawil R et al : Dementia of adult polyglucosan body disease. Evidence of cortical and subcortical dysfunction. Arch Neurol 51: 90-94, 1994.
Río-Hortega P del: El tercer elemento de los centros nerviosos. I. La microglía en estado normal. Bol Soc Esp Biol. p. 68-82, 1919.
Río-Hortega P del: La microglía y su transformación en células en bastoncito y cuerpos gránulo-adiposos. Arch Neurobiol I: 171-208, 1920.
Rio-Hortega P del: Papel de la microglía en la formación de los cuerpos amiláceos del tejido nervioso. Bol R Soc Esp Hist Nat XXV: 127-141, 1925.
Río-Hortega P del: El maestro y yo. C.S.I.C. Madrid 1986.
Robertson NP, Wharton S, Anderson J, Scolding NJ: Adult polyglucosan body disease associated with an extrapiramidal syndrome. J Neurol Neurosurg Psychiat 65: 788-790, 1988.
Robitaille Y, Carpenter S, Karpati G, Di Mauro S: A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes. A report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora’s disease and normal aging. Brain 103: 315-336, 1980.
Sakai M, Austin J, Witmer F, Trueb L: Studies of corpora amylacea: I. Isolation and preliminary characterization by chemical and histochemical techniques. Arch Neurol 21: 526-544, 1969.
Schroder JM, May R, Shin YS et al: Juvenile hereditary polyglucosan body disease with complete branching enzyme deficiency (type IV glycogenosis): Acta Neuropathol 85: 419-430, 1993.
Serratosa JM, Delgado Escueta AV, Posada I et al: The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum Mol Genet 4: 1657-1663, 1995.
Serratosa JM, Gomez-Garre P, Gallardo M et al: A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Human Mol Genet 8: 345-352, 1999.
Servidei S, Riepe RE, Langston C et al: Severe cardiopathy in branching enzyme deficiency. J Pediatr 111: 51-56, 1987.
Sindern E, Patzold T,Vorgerd M et al: Adulte Polyglukosan-KorperKrankheit. Fallbeispiel mit uberwiegender betiligung des zentralen und peripheren nervensystems und branchingenzymdefekt in leukozyten. Nervenarzt 70. 745-749, 1999.
Singhgrao JW, Neal JW, Newman GR: Corpora amylacea could be an indicator of neurodegeneratin. Neuropathol Appl Neurobiol 19: 269-276, 1993.
Singhrao SK, Morgan BP, Neal JW, Newman GR: Functional rol for corpora amylacea based on evidence from complement studies. Neurodegeneration 4: 335-345, 1995.
Stam FC, Wigboldus JM, Bots GThAM: Presenil Dementia. A form of Lafora disease. J Am Geriatr Soc 28: 237-240, 1980.
Sugiyama H, Hainfellner JA, Lassmann H et al: Uncommon types of polyglucosan bodies in the human brain: distribution and relation to disease. Acta Neuropathol 86: 484-490, 1993.
Suzuki K, David E, Kutschman B: Presenile dementia with «Lafora-like» intraneuronal inclusions. Arch Neurol 25: 69-80, 1971.
Thompson AJ, Swash M, Cox EL et al: Polysaccharide storage myopaty. Muscle Nerve 2: 349-355, 1988.
Virchow: Uber einige im gehirn und ruckenmark des menschen aufgefundene substanz mit der chemischen reaction der cellulose. Citado por Rio-Hortega P del: Papel de la microglía en la formación de los cuerpos amiláceos del tejido nervioso. Boletín de la Real Sociedad Española de Historia Natural XXV: 127-141, 1925.
Vos AJM, Joosten EMG, Gabreels-Festen AAWM: Adult Polyglucosan body disease: clinical and nerve biopsy findings in two cases. Ann Neurol 13: 440-444, 1983.
Wang J, Stuckey JA, Wishart MJ, Dixon JE: A unique carbohydrate binding domain targets the Lafora disease phosphatase to glycogen. J Biol Chem 277: 2377-2380, 2002.
Weis J, Schroder JM: Adult polyglucosan body myopathy with subclinical neuropathy: Case report and review of diseases associated with polyglucosan body accumulation. Clin Neuropathol 7: 271-279, 1988.
Wilson JD, Horoupian DS: Bielschowsky bodies (Lafora bodies of Bielschowsky type): report of a case associated with Rosenthal fibers in the brain stem. Acta Neuropathol 102: 505-509, 2001.
Wisniewski T, Goldman JE, Resor L: Dementia associated with polyglucosan bodies (Abstract). Neurology 40 (Suppl 1): 338, 1990.
Yokota T, Ishihara T, Kawano H et al: Immunological homogeneity of Lafora body, corpora amylacea, basophilic degeneration in heart, and intracytoplasmic inclusions of liver and heart in type IV glycogenosis. Acta Pathol Jpn 37: 941-946, 1987.
Ziemssen F, Sindern E, Schroder JM et al: Novel missense mutations in the glycogen-branching enzyme gene in adult polyglucosan body disease. Ann Neurol 47: 536-540, 2000.
![]()