
Vol. 35, n.º 4, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 4, 2002 |
A. Martínez1, A. Rábano2
1 Servicio de Anatomía Patológica, Sección de Neuropatología, Hospital Clínico de San Carlos, Madrid. 2 Laboratorio de Neuropatología, Fundación Hospital Alcorcón y Banco de Tejidos para Investigación Neurológica, Universidad Complutense, Madrid.
RESUMEN
La Enfermedad de Huntington (EH) es un proceso neurodegenerativo que afecta fundamentalmente al estriado, provocando trastornos motores (corea, rigidez), cognitivos y psiquiátricos de carácter progresivo, fundamentalmente en la edad adulta. Se hereda con carácter autosómico dominante debido a una mutación en el gen de la huntingtina (Htt), proteína de función desconocida. La mutación consiste en un aumento del número de tripletes CAG que codifican para una secuencia de poliglutamina en el N-terminal de la Htt.
Desde el punto de vista neuropatológico, la enfermedad EH se caracteriza básicamente por dos hechos: 1) pérdida de neuronas y gliosis que afecta fundamentalmente al estriado (despoblación selectiva de neuronas medianas que produce atrofia del caudado-putamen) y, en menor medida a la corteza cerebral (pérdida de células piramidales de proyección). 2) Agregación de Htt mutada en forma de cuerpos de inclusión intranucleares y de neuritas distróficas, detectables inmunocitoquímicamente en las neuronas de los territorios afectados.
La EH es el prototipo de proceso en el que los avances en patología celular y genética molecular, con la creación de animales transgénicos y líneas de células transfectadas, junto al aprovechamiento racional de cerebros con este proceso procedentes de autopsias en Bancos de Cerebros, ha permitido un avance considerable en el conocimiento de la patogenia de esta enfermedad y otras enfermedades neurodegenerativas.
INTRODUCCIÓN
En 1872 George Huntington (1850-1916) hizo la primera descripción clínica completa y clara de una enfermedad familiar, cuyos pacientes había estudiado junto a su abuelo y su padre en Long Island, Nueva York. El seguimiento familiar de los afectados condujo posteriormente hasta dos hermanos, que en 1630 partieron con sus familias desde Essex (SE de Inglaterra) hacia Boston. En los tres siglos siguientes, unos 1.000 descendientes padecieron la enfermedad; muchos de ellos fueron acusados de brujería, al ser interpretados sus movimientos anormales como «burla a Jesucristo en la cruz» (1).
La Enfermedad de Huntington (EH) afecta a todas las razas, con una incidencia de 4-7/100.000 habitantes en los paises occidentales, siendo mucho más baja en individuos de raza negra, orientales y finlandeses. Hay, sin embargo, grupos con mayor incidencia en la región de Zulia (NO de Venezuela, junto al lago Maracaibo), en la isla de Tasmania (Sur de Australia) y en Moray Firth (estuario en el mar del Norte, al NE de Escocia) (2,3).
Aunque su edad media de presentación se sitúa en torno a los 40 años, se puede manifestar desde la infancia a la vejez (2,4). La forma del adulto, coreica y demenciante, es la más frecuente. Cursa con movimientos coreoatetósicos (espasmódicos, impredecibles, involuntarios y constantes), frecuentemente asociados a alteraciones psiquiátricas y cognitivas. El curso es progresivo, con una evolución media de unos 15 años hasta la muerte, que ocurre por enfermedad intercurrente, si bien existe alto riesgo de suicidio. La forma infantil, rígida, es menos frecuente y suele cursar con convulsiones y un cuadro rígido-acinético; el curso suele ser más rápido que el del padre (que es el progenitor que suele transmitir la enfermedad en estas formas precoces).
La EH es una enfermedad hereditaria de tipo autosómico dominante, con penetración completa y fenómeno de anticipación. El gen responsable, localizado en el brazo corto del cromosoma 4 (4p16.3), fué caracterizado en 1993 y denominado IT15 (2-4). Tiene una amplia expresión, no sólo en el sistema nervioso (neuronas y glía), sino tambien en gran variedad de tejidos (5). Este gen codifica una proteína denominada huntingtina (Htt), vital la embriogénesis del SN pero de la que no se conoce una función específica (6,7). La mutación radica en una expansión de repeticiones de tripletes CAG (citosina-adenina-guanina), que codifican una región poliglutamínica (poliQ) en el extremo N-terminal de la Htt. El gen normal contiene entre 9 y 35 copias, mientras que en los genes que causan la enfermedad el número de repeticiones de tripletes es superior a 36-40 (2-4,8). Se trata de una de las al menos 10 enfermedades hereditarias autosómicas dominantes producidas por expansión excesiva de tripletes CAG en sus respectivas proteínas (7,9,10) (cuadro 1). En todas ellas el único factor que comparten sus proteínas específicas es la expansión de las repeticiones de tripletes CAG, que se traducen en una expansión poliQ. Cuanto mayor es el número de repeticiones antes se manifiesta el proceso, lleva curso clínico más grave y las alteraciones neuropatológicas son más severas (3,8,11). La longitud de la expansión de trinucleótidos es inestable, de forma que los descendientes tienen diferente número de repeticiones que los progenitores (6,12). Además, en cada enfermo puede haber diferencias en la longitud de la expansión CAG según el tejido considerado (mosaicismo). El tamaño de las expansiones es particularmente inestable en los espermatozoides, y probablemente la meiosis repercute en gran medida en su inestabilidad, provocando aumento del número de repeticiones CAG, lo que explica que la transmisión paterna sea la que con mayor frecuencia provoca el fenómeno de la anticipación. Dentro del propio SNC la longitud de las expansiones es diferente según la región considerada; así, en estriado y corteza cerebral las expansiones CAG son más largas que en el cortex cerebeloso.

ANATOMÍA PATOLÓGICA
El presente trabajo está basado en el estudio de cerebros de autopsia con EH, existentes en el Banco de Tejidos para Investigación Neurológica de Madrid (13 casos, 12 de adultos y uno infantil), procedentes de donaciones de familiares y remitidos, bien directamente al Banco, bien a través de los Servicios de Anatomía Patológica de los Hospitales en los que se realizó la autopsia.
Al contrario que otras enfermedades neurodegenerativas, hasta muy recientemente no se conocían marcadores histológicos definidos de la EH y durante muchos años ha sido considerada como una encefalitis, al atribuir a cuadros infecciosos secundarios o concomitantes la base patológica primaria del proceso. Quizá por esta razón, su neuropatología tardó en ser establecida de forma precisa. Bielschowsky (1922) atribuye a Jelgersma (1908) el primer estudio patológico válido de la EH y la clara implicación del STR, y del CDD en particular, en este proceso (13), hallazgos que fueron confirmados por Alzheimer (1911) y otros (13,14). El propio Bielschowsky (1922) llamó la atención sobre la conservación de las neuronas de gran tamaño del estriado (15). Dunlap (1927) realizó contaje de neuronas en los núcleos estriado, encontrando lesiones más intensas en las zonas media y posterior del putamen (14). Dom y col (1973 y 1976) cuantificaron la proporción entre neuronas pequeñas y grandes, cuya relación normal de 145-175/1 se reducía a 26-40/1 (15).
De este modo quedaron establecidas las bases del diagnóstico morfológico de la EH, según criterios topográficos (afectación del neoestriado) y de selectividad en la afectación neuronal (pérdida de neuronas de pequeño tamaño). El aislamiento del gen de la EH y de su producto génico (la proteina Htt), junto con el desarrollo de anticuerpos frente a la Htt normal y mutada, que posibilitó la demostración agregados proteicos intraneuronales en ratones transgénicos, en líneas celulares transfectadas y en cerebros pacientes con EH (16-18), han proporcionado un marcador morfológico específico de esta enfermedad y otras producidas por expansiones poliQ (19).
Un paso importante para el conocimiento de la evolución de las lesiones en la EH y los subtipos neuronales afectados y su correlación anatomoclínica y fisiopatológica, ha sido la centralización del estudio de encéfalos procedentes de necropsia de enfermos con esta enfermedad (20).
Un primer estudio permitió establecer un sistema de gradación de las lesiones (21), basado en el grado de atrofia del estriado. Así, se establecieron 5 grados de severidad macroscópica (G.0 a 4) que se correlacionan con el tiempo de evolución de la enfermedad, la gravedad clínica y el grado de pérdida neuronal y gliosis del estriado. Este sistema sigue utilizándose en la actualidad (2-4) (cuadro 2). La atrofia macroscópica se valora por la impronta que el perfil medial de la cabeza del caudado imprime en al asta frontal del ventrículo lateral. En los grados 0 y 1 no se objetivan alteraciones macroscópicas; microscópicamente, aunque en el G.0 ya existe pérdida neuronal importante (30-40%), sólo es apreciable mediante estudios cuantitativos y no existe gliosis reactiva; en el grado 1 la pérdida neuronal es leve con las técnicas de estudio habituales (50% tras contaje de neuronas), pero ya se aprecia gliosis en el caudado. En los grados 2, 3 y 4, la atrofia de la cabeza del caudado del rectifica progresivamente su perfil medial (ventricular), de forma que primero pierde cierto grado de convexidad (G.2), luego se hace recto (G.3) y finalmente cóncavo (G.4) (fig. 1).

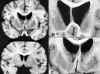
Fig. 1. Alteraciones macroscópicas en la enfermedad de Huntington. A)
Cortes sagitales correspondientes a un cerebro con EH (imagen superior) y a un
control normal (corte inferior). En la EH se aprecia atrofia de la cabeza del
núcleo caudado, que muestra un perfil medial recto en vez del convexo normal,
provocando dilatación ventricular. También hay atrofia del putamen. B y C)
Detalles de la anterior en cortes realizados a nivel de la cabeza del caudado
(A) y del lenticular (B):
Paralelamente, aumenta el grado de lesión microscópica, que se desarrolla según un gradiente topográficamente ordenado, en sentido medio-lateral, póstero-anterior y caudo-rostral (20, 21), de forma que la cola del caudado se afecta antes que el cuerpo, éste antes que la cabeza y la parte medial (periventricular) antes que la lateral. El mismo fenómeo ocurre en el putamen. La pérdida neuronal se acompaña de gliosis progresiva, que avanza según el mismo gradiente (fig. 2). El G.3 es el más frecuente, seguido del G.2 y el G.4; sin embargo, en la forma juvenil es más frecuente el G.4.

Fig. 2. Microfotografías del
caudado en un caso de EH avanzado, teñidas con hematoxilina-eosina (A) y con
técnica inmunohistoquímica para proteína gliofibrilar (B). Con HE (A) se
observa pérdida de la arquitectura normal del núcleo, con despoblación
neuronal y aspecto fibrilar tosco del neuropilo debido a la gliosis. Esta se
aprecia mejor en B (PGFA), donde se aprecian numerosos astrocitos
hipertróficos.
La afectación macroscópica no se limita al estriado. De hecho, en la mayor parte de los casos y dependiendo del tiempo de evolución de la enfermedad, siempre suele haber una atrofia global de todo el encéfalo, que suele pesar menos de 1.100 g, con cierto grado de atrofia cortical. De hecho, valoraciones cuantitativas demuestran no sólo atrofia cortical, sino también pérdida de sustancia blanca cerebral (22). También existe atrofia progresiva de pálido (sobre todo de su porción externa), la amígdala y tálamo.
La aplicación del método de Golgi y de técnicas histoquímicas (NADPH-diaforasa, acetilcolinesterasa) e inmunocitoquímicas (detección de neurotransmisores), ha permitido conocer con detalle las subpoblaciones neuronales del estriado y su afectación en la EH.
El estriado tiene 2 poblaciones neuronales principales, fácilmente identificables con el método de Nissl: una de células nerviosas de tamaño medio, que representa el 90% del total, y otra constituida por neuronas de gran tamaño. Como ya ha quedado expuesto, la EH afecta de forma selectiva el grupo neuronal más importante, el de las neuronas pequeñas-medianas de la histología clásica. Sin embargo, con el método de Golgi se pueden identificar hasta 6 tipos neuronales diferentes (23), en función del tamaño del soma, de la trayectoria del axón, la morfología general de las arborizaciones dendríticas, la cantidad de espinas y la presencia o no de varicosidades. Todos estos tipos se pueden resumir en dos: 1) neuronas espinosas, dotadas de una amplia superficie sináptica y de axón largo (neuronas de proyección), y 2) neuronas no espinosas, que establecen circuitos locales. Cajal fué el primero en describir las espinas dendríticas y un tipo de neuronas enanas que denomina «neurogliformes» (24); además, siguió el complicado trayecto axonal inicial de las neuronas espinosas medianas de axón largo, que hace que puedan confundirse con neuronas de axón corto. También señala la predilección de las colaterales de las aferencias corticales por las neuronas de axón largo (24).
Mediante estudios inmunocitoquímicos e histoquímicos se han podido relacionar los tipos neuronales demostrados con el método Golgi con las subpoblaciones neuronales según los tipos de neurotransmisores que utilizan, determinar los receptores que expresan y establecer su afectación selectiva en la EH (7,25,26,27). Las neuronas espinosas son neuronas de proyección de tamaño mediano (MSSN: medium size spiny neurons), GABAérgicas, que según los neurotransmisores que utilizan, los tipos de receptores para dopamina que expresan y la proyección de sus axones, se pueden subdividir en dos poblaciones principales: 1) Las que coexpresan encefalina y tienen receptores de dopamina tipo D2 (GPe-GABA/EnkD2); forman la vía indirecta de proyección estriatal, excitadora, a la parte externa del pálido y al núcleo subtalámico. 2) Neuronas que junto al GABA coexpresan sustancia P y dinorfina y poseen receptores de dopamina D1; constituyen la vía directa, inhibidora, y proyectan al pálido interno (GPi-GABA/Dyn/sP/D1) y a la porción reticular de la sustancia negra (SNr-GABA/Dyn/sP/D1). Entre las neuronas carentes de espinas dendríticas, hay 2 subpoblaciones fundamentales:1) neuronas de tamaño mediano, que expresan NADPHd, somatostatina y neuropéptido Y, y 2) neuronas de gran tamaño, colinérgicas (AchE+ y ChAT+).
Las MSSN son las más afectadas en la EH y muchas de ellas muestran cambios morfológicos con el método de Golgi (28,29), que han sido interpretados como el resultado de una secuencia lesional bifásica: reactiva regenerativa primero, con crecimiento dendrítico y de espinas, y regresiva después, que conduce a la muerte neuronal. La mayoría de las neuronas de proyección del estriado expresan calbindina, y son precisamente las células inmunoreactivas a esta proteína transportadora de calcio las que muestran las alteraciones dendríticas descritas con el método de Golgi (29). De las 2 subpoblaciones inmunohistoquímicas de MSSN, las GPe-GABA/EnkD2 son las primeras en degenerar y su pérdida es la responsable de los movimientos coreiformes. Las MSSN GPi-GABA/Dyn/sP/D1 y SNr-GABA/Dyn/sP/D1) desaparecen de forma más lenta y progresiva y su pérdida explica la bradicinesia (2,4,6). Las poblaciones neuronales preservadas en la EH son las interneuronas no espinosas de tamaño mediano, positivas a NADPH diaforasa (NOsintasa), somatostatina y neuropéptido Y, y las neuronas no espinosas de gran tamaño, colinérgicas, aunque también parecen degenerar en fases avanzadas del proceso (30,31). El proceso degenerativo de la EH no parece afectar a la compartimentalización del estriado en matriz/estriosomas (20), a pesar de que los primeros focos de pérdida neuronal y gliosis se detectan en los estriosomas (32), ya que, posteriormente, las lesiones afectan casi por igual a ambos compartimentos.
Aunque ha sido objeto de controversia la implicación de la corteza cerebral en la EH, debido a la dificultad de detectar anomalías en la misma (20), actualmente no existen dudas sobre de la existencia de pérdida neuronal en ciertas capas del cortex. Mediante estudios cuantitativos se ha demostrado pérdida de células piramidales de las capas III, V y VI (33-35), que no guarda relación con el grado de afectación estriatal, por lo que debe tratarse de una degeneración primaria (33); sin embargo, otros la achacan a degeneración transináptica (34,35). Algunas de las neuronas conservadas presentan aumento de la arborización dendrítica (método de Golgi) que refleja su plasticidad, en un intento de compensar la pérdida de otras células (36). En casos muy avanzados hay cierto grado de desorden arquitectural y gliosis en las capas correspondientes (fig. 3). Las neuronas de asociación local están conservadas (33).
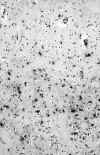
Fig. 3. Fotografía microscópica
de un corte de corteza cerebral frontal, con técnica inmunohistoquímica para
PGFA. Se observa gliosis marcada de las capas profundas del cortex (capas V-VI).
La lista de poblaciones neuronales afectadas aumenta conforme se estudian núcleos previamente no considerados (6). La atrofia progresiva del pálido, sobre todo de su porción externa, parece deberse a la disminución de las aferencias estriatales, aunque algunos han encontrado pérdida neuronal. La sustancia negra muestra retracción de la porción reticular, secundaria a pérdida de aferencias estriatales. También hay afectación severa del núcleo tuberal lateral del hipotálamo, y cambios menos importantes en algunos núcleos talámicos, núcleo subtalámico y locus cerúleo. El cerebelo muestra pérdida importante de células de Purkinje en los casos juveniles, aunque puede tener origen hipóxico en los casos que cursan con estado convulsivo.
Otra alteración encontrada de forma constante es el aumento del número de células de oligodendroglía en los G.0 y 1, pero que decrece en los G.3 y 4 (21). El aumento de oligodendroglía en portadodes presintomáticos del gen de la EH hace pensar que se trate de una alteración primaria (37). También se ha descrito activación de la microglía en las zonas afectadas (neoestriado, corteza cerebral, pálido y sustancia blanca adyacente), que es proporcional al grado de la enfermedad (38).
Aunque la especificidad topográfica de las lesiones permite hacer el diagnóstico de EH en los grados 2 a 4, con lesiones establecidas, en las fases iniciales, con lesiones leves, surgen problemas diagnósticos en aquéllos casos en los que el cuadro clínico no es concluyente y/o no se pueden constatar antecedentes familiares. La posibilidad de poner de manifiesto con técnicas inmunocitoquímicas la presencia los agregados de Htt mutada en las áreas afectadas, ha supuesto un gran avance en el diagnóstico de la EH (16-18). Estos agregados se pueden detectar con anticuerpos anti-ubicuitina y anti-expansiones poliglutamínicas (técnicas aplicables a cualquier enfermedad con expansión de tripletes CAG) y, de forma más específica, con anticuerpos anti-fragmentos N-terminales (con las expansiones poliQ) de la Htt mutada (fig. 4).
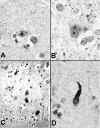
Fig. 4. Cortes histológicos de
caudado (B y C) y corteza cerebral (A y D), teñidos con técnicas
inmunocitoquímicas con anticuerpos anti-ubicutina (A, B y D) y seguimiento
N-terminal/poliQ. En A y B se observan cuerpos de inclusión intranucleares, muy
inmunoreactivos para UB, uno de gran tamaño en una neurona cortical (A) y otro
más pequeño, inmediatamente por debajo del nucleolo, en una neurona del
caudado (B)´en éste se aprecia, además, mayor tinción de fondo,
correspondiente a tinción de fibras nerviosas del neuropilo. La tinción para
el segmento poliQ muestra inmunoreactividad en numerosas neuronas del estriado
(C). D) Neurita distrófica en cortex cerebral, intensamente reactiva a UB.
La UB solo se detecta en una subclase de agregados, lo que sugiere que la proteolisis de la Htt mutada mediada por el sistema UB-proteasoma puede ser tardía e inconstante (39). También se han detectado con rojo Congo (40), lo que demuestra la capacidad de los agregados de fragmentos de proteínas anormales a sufrir cambios en su conformación, con producción de bandas-ß antiparalelas muy estables (estructura amiloide). Los agregados aparecen en las neuronas bajo dos formas: cuerpos de inclusión intranucleares y agregados citoplásmicos (paranucleares y en forma de neuritas distróficas). Su distribución se ajusta a la topografía de las lesiones ya descrita, afectando de forma preferente al neocortex y al estriado. Las inclusiones neuropílicas son más frecuentes que las nucleares (39). Las intranucleares son más numerosas en la corteza cerebral que en el estriado en todos los grados patológicos y su frecuencia guarda relación con la longitud de la expansión CAG (41) y, por tanto, con la gravedad del proceso, siendo más frecuentes en los casos juveniles. Sin embargo, el estudio de la distribución inmunohistoquímica de la Htt anómala evidencia una disociación entre el número de agregados de la misma y la distribución topográfica de las lesiones de EH por un lado, y la sensibilidad neuronal a la enfermedad, por otro (7,42). Así, todos los agregados son más frecuentes en la corteza cerebral que en el estriado, aunque éste sea el más afectado en la EH (39,42). Por otro lado, estudios con doble tinción en el estriado muestran que hay más inclusiones en las interneuronas NADPHd resistentes (50% de neuronas con inclusiones) que en las neuronas calbindina-positivas vulnerables (4% de inclusiones) (42). En la corteza cerebral la situación es distinta, pués hay inclusiones intranucleares en más del 90% de las neuronas piramidales (sensibles), mientras que son mucho menos frecuentes en las interneuronas (resistentes) (7). Estos hallazgos parecen demostrar que la agregación de fragmentos N-terminales de Htt mutada puede ser un mecanismo citoprotector contra neurotoxicidad poliQ (42). Otro hallazgo importante ha sido la demostración de agregados proteicos (intranucleares y neuropílicos) en portadores presintomáticos (39, 37), incluso muchos años antes del comienzo inicio clínico de la enfermedad, según la ecuación predictiva de Duyao (37).
PATOGENIA
La EH es una de las 10 enfermedades producidas por expansión de tripletes de nucleótidos CAG, que se traduce en una expansión poliglutamínica en el N-terminal de una proteína, específica para cada proceso (6,9,10,12,43,44). El único dato que comparten estas enfermedades es la expansión de las repeticiones poliQ en el N-terminal de las proteínas correspondientes, las cuales, por lo demás, tienen funciones diferentes, son codificadas por genes que están muy dispersos en el genoma y su efecto patógeno presenta una gran selectividad topográfica, aunque con cierto solapamiento (6). Sin embargo, comparten una serie de hechos comunes. El efecto que la expansión poliQ produce en todas ellas, más que una pérdida o disminución de su función normal, es una ganancia de función de carácter tóxico (6,12,43), aunque algunos aspectos se explican también por pérdida de función (12,45).
En todas estas enfermedades existe un defecto en la degradación de la proteína mutada, que da lugar a la formación de fragmentos N-terminales con las expansiones poliQ que tienen tendencia a la agregación, dando lugar a la formación de inclusiones intracelulares en los territorios afectados (6,7,46,47). La alteración de la degradación de las proteínas mutadas es fundamental en la patogenia de las lesiones, pués son las propiedades de los fragmentos N-terminales (con las expansiones poliQ) las responsables de la toxicidad celular (7,46-50). Tanto la Htt normal como la mutada son degragadas por el proteasoma, pero la velocidad de la degradación es inversamente proporcional a la longitud de las expansiones poliQ (49). Se ha demostrado que la inhibición de actividad del sistema UB-proteasoma duplica la cantidad de agregados ubicuitinados, indicando que las inclusiones intracelulares se forman cuando se agota la capacidad de dicho sistema para degradar Htt mutada, propensa a la agregación (50). El defecto de la degradación de la Htt puede ocurrir por sobrecarga de proteína anormal, que podría sobrepasar la capacidad degradativa del proteasoma, o por disminución de su función como consecuencia de la enfermedad, hecho que ha sido probado (48). La presencia de ubicuitina en los agregados debe representar un fallo celular para remover proteínas anormales, fallo que permite su agregación (48). La UB puede tener efecto protector: sus propiedades físicas hacen que el recubrimiento por ésta impida mayor agregación, evitando además interacciones con otras proteínas que podrían desencadenar mecanismos dañinos para la célula (48).
Pero todos estos mecanismos, que parecen ser comunes a este grupo de enfermedades, no explican la selectividad regional de las lesiones. Uno de los hechos que primero se ha investigado es la intensidad de la expresión génica en diferentes regiones. Así, la HTT se expresa en todas las neuronas del SNC (5,43) y en mumerosas células somáticas (5,11). Sin embargo, el mosaicismo hace que la longitud de las expansiones CAG varíe en diferentes tejidos, e incluso en diferentes estructuras dentro del tejido nervioso: las expansiones son más cortas en el cerebelo que en la corteza frontal, el estriado y otras regiones, predominando siempre la expresión neuronal sobre la glial (5,11). Sin embargo, las diferencias regionales no son muy grandes y no existe correlación entre la diferencia de longitud de las repeticiones de CAG en diferentes regiones del SNC y los niveles del transcripto con la vulnerabilidad regional (5,6). Además, ya se ha señalado que el número de inclusiones celulares tampoco guarda relación con la vulnerabilidad de los territorios afectados (39,42). Por tanto, la selectividad topográfica lesional debe tener otras explicaciones y, tanto en cerebros humanos con EH como en ratones transgénicos y en cultivos de líneas celulares transfectadas, se han encontrado otros factores que pueden influir en la vulnerabilidad selectiva.
Uno de los hechos que podrían explicar en parte la distribución de las lesiones es la diferencia en el patrón proteolítico, junto con una ubicuitinización también específica de ciertos fragmentos N-terminales, en el estriado con respecto de la corteza cerebral (47). Esta diferencia puede implicar que los mecanismos de reconocimiento y eliminación de las proteína mutada sean distintos en diferentes tipos celulares, y que neuronas con sistemas más eficientes sean más resistentes a la lesión (7).
Otra propiedad importante de las expansiones poliQ es su capacidad para reclutar e interaccionar con otras proteínas (46) y su efecto sobre la transcripción génica, pudiendo simular la acción de ciertos factores de transcripción con dominios ricos en glutaminas, como la proteína TBP, uniéndose directamente al DNA y produciendo represión o subregulación de la transcripción de ciertos genes, como la expresión de receptores de neurotransmisores (7). En este sentido, es importante la demostración en líneas celulares que coexpresan HTT mutada y dos subclases de receptores glutamínicos (NR1A/NR2A o NR1A/NR2B), de una susceptibilidad diferente a excitotoxicidad in vitro. Las células que coexpresan mHTT-138Q y receptores tipo NR1A/NR2B son mucho más sensibles y muestran además fenómenos apoptóticos; como el NR1A/NR2B es el subtipo predominante de receptor de glutamato en las MSSN, debe ser éste un factor importante en la neurodegeneración selectiva (51). Esta observación viene a confirmar la teoría de la excitotoxicidad como causante de la muerte de las neuronas estriatales, mediante estimulación de baja intensidad, pero mantenida, de la vía córtico-estriatal glutaminérgica sobre las MSSN, previamente dañadas por la mutación.
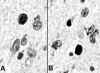
Fig. 5. Microfotografías del
caudado teñido con técnica TUNEL para demostración de apoptosis. Se observa
inmunoreacción en varios núcleos, con imágenes de apoptosis en diferentes
períodos evolutivos.
La causa de la muerte neuronal implica a varios factores (aparte de la excitotoxicidad), como el estrés oxidativo, la insuficiencia energética (existe disminución de la actividad de succinato y COX y alteración de la fosforilación exidativa) (43), disminución de la producción de BDNF (45) y de su expresión en el estriado (52), alteración del potencial de membrana mitocondrial, con liberación de citocromo-C al citosol y activación de las caspasas-9 y 3 (49) y el aumento de la expression de caspasa-1, que a su vez puede activar la 3 y desencadenar APP (53,54). Las caspasas están muy implicadas en la EH pues, además de intervenir en la partición directa de la Htt, generando fragmentos con poliQ tóxicos, pueden ser reclutadas y activadas por los agregados con poliQ (55). Estos factores y sobre todo la activación de las caspasas desencadenan apoptosis (fig. 5) y necrosis en las neuronas susceptibles (56).
Todos estos acontecimientos requieren la expresión continuada de la proteina mutada: el desarrollo de ratones transgénicos con expresión condicionada, mediada por tetraciclinas, del gen de la EH, ha demostrado que el bloqueo de la expresión hace desaparecer las inclusiones de agregados proteicos patológicos y mejora la clínica (57).
AGRADECIMIENTOS: Deseamos expresar nuestro sincero agradecimiento a los pacientes con Enfermedad de Huntington donantes del Banco de Tejidos para Investigación Neurológica, a sus familiares, y a los neurólogos y patólogos que han contribuido a la incorporación de tejido de Enfermedad de Huntington al archivo del BTIN desde los siguientes hospitales: Fundación Jiménez Díaz (Madrid), H. Arnau de Vilanova (Lleida), H. General U. Gregorio Marañón (Madrid), H. Virgen de la Salud (Toledo), H. Ramón y Cajal (Madrid), H. Sondureta (Bilbao), H. de Laredo (Laredo). H. San Telmo (Palencia), H. Gral. Yagüe (Burgos) y H. del Niño Jesús (Madrid).
BIBLIOGRAFÍA
Arbor A, DeJong RN. George Huntington (1850-1916). En The Founders of Neurology, ed por W Haymaker W y F Schiller. Springfield, Illinois: Charles C Thomas Pub, 2.ª ed, 1970: 453-6.
Lowe J, Lennox G, Leigh PN. Disorders of movement and system degenerations. En: Greenfield’s Neuropathology, V. 2, ed por DI Graham y PL Lantos. Londres: Arnold, 6.ª ed., 1997: 281-366.
Azzarelli A. Enfermedad de Huntington y degeneraciones de algunos núcleos subcorticales. En Neuropathologia, diagnóstico y clínica, ed por FF Cruz Sanchez. Barcelona: Edimsa, 2000: 603-20
Giordana MT. Huntington’s disease. En Neuropathological Diagnostic criteria for Brain Banking, ed por FF Cruz-Sánchez, R David y ML Cuzner. Amsterdam: IOS Press, 1995: 89-98.
Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, Kaatz KW, Collins FS, Albin RL. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat Genet 1993; 5:259-65.
Robitaille Y, Lopes-Cendes I, Becher M, Rouleau G, Clarck AW. The neuropathology of CAG repeat diseases: review and update of genetic and molecular features. Brain Pathol 1997; 7: 901-26.
Sieradzan KA, Mann DM. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol Appl Neurobiol 2001; 27: 1-21.
Penney JB Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol 1997; 41: 689-92.
Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 2001; 29:377-8.
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 2001; 10: 1441-8.
Furtado S, Suchowersky O, Rewcastle B, Graham L, Klimek ML, Garber A. Relationship between trinucleotide repeats and neuropathological changes in Huntington’s disease. Ann Neurol 1996; 39: 132-6.
La Spada AR, Clarck AW. Inherited neurodegenerative disorders caused by CAG/polyglutamine tract expansions: symposium introduction. Brain Pathol 1997; 7: 877-880.
Bruyn GW. Huntington’s chorea: historical, clinical and laboratory synopsis. En Handbook of Clinical Neurology, V. 6: Diseases of the basal ganglia, ed por PJ Vinken y GW Bruyn. Amsterdam: North-Holland Pb Co, 1968: 298-378.
Dreese MJ, Netsky MG. Degenerative disorders of the basal ganglia. En Pathology of the Nervous System, V.1, ed por J Minckler. New York: McGraw-Hill Book Co, 1968: 1185-1204.
Roos RAC. Neuropathology of Huntington’s chorea. En Handbook of Clinical Neurology, V. 49: Extrapyramidal disorders, ed por PJ Vinken, GW Bruyn y HL Klawans, Amsterdam: Eslsevier Sci Pub, 1986: 315-326.
Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, Weber C, Agid Y, Hirsch EC, Mandel JL. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat Genet 1995; 10: 104-10.
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intraneuronal inclusions underlies the neurological dysfunction in mice transgenic mice for the HD mutation. Cell 1997; 90: 537-48.
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277: 1990-3.
Davies SW, Beardsall K, Turmaine M, DiFiglia M, Aronin N & Bates GP. Are neuronal intranuclear inclusions the common neuropathology of triplet-repeat disorders with polyglutamine-repeat expansions? Lancet 1998; 351: 131-3.
Richardson EP. Third Dorothy S. Russell memorial lecture. Huntington’s disease: some recent neuropathological studies. Neuropathol Appl Neurobiol 1990; 16: 451-60.
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985; 44: 559-577.
De la Monte SM, Vonsattel JP, Richardson EP. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J Neurpathol Exp Neurol 1988; 47: 516-525.
Pasik P, Pasik T & DiFiglia M. The internal organization of the Neostriatum in mammals. En: The Striatum, ed por J Divac y RG Oberg. New York: Pergamon Press, 1979: 5-36.
Ramón y Cajal S. Corp strié. En Histologie du Système Nerveux de l’homme et des vertebrés, V. 2. Madrid: CSIC. 1909 (reed. 1955): 504-18.
Graybiel AM, Penney JB. Chemical architecture of the basal ganglia. En Handbook of Chemical Neuroanatomy, V. 15: The primate Nervous System, ed. por FE Bloom, A BJörklund y T Hökflt. Londres: Elsevier, 1999: 227-84.
Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 2000; 97: 505-19.
Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington’s disease. Brain Res Brain Res Rev 2000; 34: 80-101.
Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985; 227: 770-3.
Ferrante RJ, Kowall NW, Richardson EP Jr. Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci 1991; 11: 3877-87.
Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP Jr. Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J Neuropathol Exp Neurol 1987; 46: 12-27.
Ferrante RJ & Kowall NW. Tyrosine hydroxylase-like immunoreactivity is distributed in the matrix compartment of normal human and Huntington’s disease striatum. Brain Res 1987, 416: 141-6.
Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington’s disease. J Neuropathol Exp Neurol 1995; 54: 105-20.
Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington’s disease cortex. Ann Neurol 1990; 27: 200-4.
Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci Lett 1991; 133: 257-61.
Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH. Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology 1991; 41: 1117-23.
Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington’s disease: a quantitative Golgi study. Neurology 1993; 43: 2088-96.
Gomez-Tortosa E, MacDonald ME, Friend JC, Taylor SA, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel JP, Myers RH. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol 2001; 49: 29-34.
Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 2001; 60: 161-72.
Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci 1999; 9: 2522-34.
McGowan DP, van Roon-Mom W, Holloway H, Bates GP, Mangiarini L, Cooper GJ, Faull RL, Snell RG. Amyloid-like inclusions in Huntington’s disease. Neuroscience 2000; 100: 677-80.
Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 1998; 4: 387-97.
Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol 1999; 46: 842-9.
Ross CA, Becher MW, Colomer V, Engelender S, Wood JD, Sharp AH. Huntington’s disease and dentatorubral-pallidoluysian atrophy: proteins, pathogenesis and pathology. Brain Pathol 1997; 7: 1003-1016.
Margolis RL, O’Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, Callahan C, Hwang HS, Troncoso JC, Ross CA. A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol 2001; 50: 373-80.
Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001; 293: 493-8.
Evert BO, Wullner U, Klockgether T. Cell death in polyglutamine diseases. Cell Tissue Res 2000; 301(1): 189-204.
Mende-Mueller LM, Toneff T, Hwang SR, Chesselet MF, Hook VY. Tissue-specific proteolysis of Huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington’s disease striatum. J Neurosci 2001; 21: 1830-7.
Gray DA. Damage control: a possible non-proteolytic role for ubiquitin in limiting neurodegeneration. Neuropathol Appl Neurobiol 2001; 27: 89-94.
Jana NR, Zemskov EA, Wang Gh, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet 2001; 10: 1049-59.
Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 2001; 12: 1393-407.
Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci 2001; 17: 41-53.
Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res 2000; 866: 257-61.
Li SH, Lam S, Cheng AL, Li XJ. Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis. Hum Mol Genet 2000; 9: 2859-67.
Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature 1999; 399: 263-7.
Wellington CL, Hayden MR. Caspases and neurodegeneration: on the cutting edge of new therapeutic approaches. Clin Genet 2000; 57: 1-10.
Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci 1995; 15: 3775-87.
Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000; 101: 57-66.
![]()