
Vol. 35, n.º 4, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 4, 2002 |
Javier Figols Ladrón de Guevara1, Juan Carlos García-Moncó2, José Miguel Polo Esteban2 Andrés González-Mandly3, J. Fernando Val-Vernal1
Departamentos de Anatomía Patológica1
y Radiodiagnóstico3 del Hospital Universitario «Marqués de
Valdecilla» y Facultad de Medicina de la Universidad de Cantabria, Santander.
Servicio de Neurología2 del Hospital Galdakao de Vizcaya.
HISTORIA CLINICA
Mujer de 57 años enfermera de profesión, que ingresó para estudio por trastorno de la marcha. Falleció tres meses más tarde.
Antecedentes: Fumadora hasta 15 años antes. Nódulo pulmonar solitario en RX de tórax que en la TAC torácica resultó poco definido, por lo que se le sometió a un PET oncológico cérvico-pélvico que fue normal. Histerectomía por miomas .Un mes antes del ingreso había comenzado con pesadez y dolorimiento en las piernas. En los días siguientes se quejó de dificultad para caminar, con desviación hacia la derecha. Una evaluación neurológica a los diez días del inicio de estos síntomas fue prácticamente normal, aunque con hiperreflexia miotática generalizada. Progresivo empeoramiento con creciente inestabilidad, cambio de carácter, apatía y retraimiento: se encontraba depresiva y preocupada por su salud. En los días previos al ingreso presentó urgencias miccionales y, de forma ocasional, trastorno del lenguaje, dando la impresión que no encontraba algunas palabras. Buen estado general, no cefaleas, trastorno visual ni otros síntomas. Estaba en tratamiento con ansiolíticos. Ante esta sintomatología se decidió su ingreso en el hospital.
A su ingreso, la temperatura axilar, la tensión y el pulso fueron normales. Se encontraba despierta y orientada. Lenguaje normal, aunque poco fluido con contestaciones lentas. Capacidad de juicio, memoria y cálculo, normales. No apraxia de extremidades. Pobre expresividad facial. Marcha torpe y lenta, arrastrando los pies, con inestabilidad en los giros. Hipertonía plástica en extremidades superiores. Fuerza muscular normal. Reflejos miotáticos exaltados (+++), con respuestas plantares extensoras.
Analítica: Hemograma, bioquímica hemática, marcadores tumorales, perfil lipídico, proteinograma, perfil tiroideo, así como bioquímica y sedimento urinarios fueron normales .Serologías frente a lúes, VIH, borrelia y brucella, negativas. ECG normal, Rx de tórax no valorable por escasa colaboración.
Estudio radiológico (Dr. González Mandly): En la TAC craneal (figs. 1 y 2) se observaba una amplia hipodensidad que afectaba al cuerpo calloso y a la sustancia blanca subcortical de ambos hemisferios, con borramiento de surcos corticales y sin modificaciones tras la administración de contraste.
![]()
Figs. 1 y 2.
En la Resonancia Magnética encefálica (figs. 3 y 4) se observaba en secuencias T1 engrosamiento del cuerpo calloso con reducción de la señal en el mismo. La sustancia blanca de los centros semiovales mostraba realce.
![]()
Figs. 3 y 4. RMN: Engrosamiento del
cuerpo calloso.
En secuencias FLAIR (figs. 5 y 6) y T2, llamaba la atención el incremento de la señal de la sustancia blanca en el cuerpo calloso y centros semiovales que se extendía por el brazo posterior de ambas cápsulas internas llegando hasta la calota mesencefálica.
![]()
Figs. 5 y 6.
La RMN espinal era normal.
LCR: claro y transparente. Contenía 3 células /ml, con proteínas de 49 md/dl y glucosa de 55mg/ml (glucemia simultánea de 92 mg/dl), sin bandas oligoclonales ni síntesis intratecal de IgG; la proteína 14.3.3 fue negativa. El estudio microbiológico licuoral fue negativo incluyendo cultivo para bacterias y hongos, e investigación de anticuerpos frente a virus-ADN y borrelia, así como el estudio con PCR de polioma-virus (virus JC, BK y SV-40), enterovirus y herpesvirus. El examen citológico del líquido se interpretó como hipocelular con linfocitosis.
En el EEG, el ritmo de fondo estaba globalmente enlentecido, observándose brotes de ondas lentas de morfología irregular, ocasionalmente trifásicas, de hasta 120-150 mV de amplitud, proyectadas en ambos hemisferios con mayor voltaje en regiones anteriores. El estudio electromiográfico y el de la conducción nerviosa periférica fueron normales. Evolución clínica.- En los primeros días de estancia en el hospital se produjo un rápido empeoramiento clínico. Se mostraba muy distraída, tardaba en comprender y responder y precisaba de ayuda para incorporarse y dar unos pasos. Incontinencia urinaria. Un tratamiento inicial con Sinemet fue suspendido a los pocos días ante la falta de mejoría. Posteriormente se le administraron por vía oral 4 mg de metilprednisolona cada 8 horas. Al cabo de pocos días era incapaz de leer y permanecía quieta y en silencio, con ocasionales accesos de risa espasmódica; su conversación era escasa y en ocasiones parecía no comprender nada. La marcha se hizo imposible por incapacidad de mantenerse en bipedestación. A la semana del ingreso era incapaz de manejar los utensilios de comida, precisando ayuda para cualquier actividad elemental. Tres semanas después del ingreso le fue practicada una biopsia del lóbulo frontal derecho mediante técnica esterotáxica (figs. 7 y 8), que mostró gliosis astrocitaria de carácter inespecífico en sustancia blanca cerebral. La biopsia no incluyó corteza.
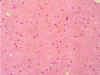
Fig. 7. HE.

Fig. 8. GFAP.
Veinte días más tarde, una nueva biopsia en región parietal posterior derecha confirmaba la existencia de gliosis astrocitaria observada en la biopsia anterior, en esta ocasión asociada a intensa espongiosis con vacuolas multitabicadas (fig. 9); la inmunohistoquímica para PrP fue negativa.

Fig. 9.
A partir de este momento su nivel de relación con el entorno se fue reduciendo progresivamente; pasaba el tiempo sentada o encamada sin emitir sonidos ni realizar movimientos espontáneos, aunque ocasionalmente seguía con la mirada. A punto de cumplirse el segundo mes de estancia hospitalaria, se observó hemiparesia derecha progresiva. No presentaba mioclonías ni otros movimientos involuntarios. Varios EEGs seriados señalaban un progresivo deterioro de la actividad cerebral, con cierta acentuación en hemisferio izquierdo. En un polisomnograma se observó ausencia del patrón normal del sueño, ausencia de sueño REM y alternancia de fases de actividad theta de mediano voltaje con otras de actividad theta-delta de mayor voltaje y ondas de morfología trifásica, que en aislados brotes de hasta 10 segundos, tenían tendencia a la pseudoperiodicidad. En una RMN encefálica practicada transcurrido un mes desde la anterior, se observaron lesiones similares a la primera. El tratamiento esteroideo se redujo hasta suspenderlo. Al final del segundo mes de estancia desarrolló tromboflebitis en la extremidad inferior izquierda y se instauró anticoagulación. Presentó fiebre inconstante.Los síntomas neurológicos no cambiaron. Al inicio del tercer mes sufrió repetidos episodios sépticos de origen respiratorio y urinario. Falleció cuatro meses después del inicio de los síntomas neurológicos. Se practicó estudio necrópsico restringido al sistema nervioso central intracraneal.
DISCUSION CLÍNICA-DIAGNOSTICO DIFERENCIAL (Dr. García-Moncó)
En síntesis, se trata de una paciente con una enfermedad neurológica progresiva que le condujo al exitus en 4 meses. Clínicamente, presentaba un deterioro cognitivo y un síndrome piramidal y extrapiramidal, lo que sugiere una enfermedad difusa del sistema nervioso central. El déficit cognitivo es consistente con una demencia subcortical, entidad dominada por la bradicinesia, la apatía y la falta de iniciativa que condujo al mutismo. Este síndrome es similar al que ocurre con lesiones frontales, y probablemente se debe a que las lesiones de sustancia blanca producen una desconexión de los lóbulos frontales con diversas estructuras subcorticales. A diferencia del clásico síndrome frontal, están ausentes los signos «frontales puros». Tampoco se produce el síndrome de afasia, apraxia y agnosia que acaba dominando las demencias corticales. Consistente con su cuadro clínico, la resonancia magnética cerebral mostraba una alteración difusa de toda la sustancia blanca cerebral de ambos hemisferios, junto a un notable engrosamiento del cuerpo calloso. No había captación de contraste parenquimatosa ni leptomeníngea, presencia de masas tumorales ni aparente afectación de la sustancia gris. Finalmente, se practicaron dos biopsias cerebrales que mostraron diferentes grados de espongiosis.
La afectación difusa y adquirida de la sustancia blanca cerebral en el adulto obliga a un amplio diagnóstico diferencial debido a la diversidad de su etiología (tabla I). En esta discusión revisaremos las más pertinentes al cuadro clínico de la paciente.

En primer lugar, las enfermedades desmielinizantes, paradigma de las cuales es la esclerosis múltiple, ocasionalmente producen una afectación tan extensa de la sustancia blanca. En esta paciente, sin embargo, la evolución fue extremadamente agresiva y maligna, algo atípico para la forma clásica de esta enfermedad, y que plantearía el diagnóstico de la variante de Marburg, una leucoencefalitis hemorrágica (Hurst) o una encefalitis aguda diseminada. Sin embargo, la neuroimagen, aunque no las descarta, no es característica de estas entidades, el líquido cefalorraquídeo no mostraba pleocitosis ni síntesis de inmunoglobulinas o bandas oligoclonales; algunas de estas anomalías hubieran sido esperables en una enfermedad desmielinizante. Por último, la biopsia no fue consistente. Diversas enfermedades sistémicas de carácter inflamatorio pueden conducir a una afectación de la sustancia blanca cerebral, como el lupus o la sarcoidosis, entre otras. Sin embargo, ninguna de estas entidades estaba presente en esta paciente, el LCR era normal, no había captación meníngea y la evolución clínica no era consistente.
La isquemia cerebral de larga evolución puede involucrar preferentemente a la sustancia blanca, dando lugar a la encefalopatía de Binswanger, un cuadro de lenta evolución asociado a hipertensión arterial. La ausencia de hipertensión arterial así como la evolución maligna en este caso y el engrosamiento del cuerpo calloso no son consistentes con estas entidades. Las vasculitis que afectan al sistema nervioso pueden cursar también con leucopatía, y pueden estar confinadas exclusivamente al sistema nervioso (vasculitis granulomatosa aislada al SNC) o encuadrarse dentro de una enfermedad sistémica. En esta paciente la ausencia de patología sistémica asociada, la normalidad del LCR (las vasculitis suelen cursar con pleocitosis), la extensísima leucopatía —inusual en una vasculitis—, el engrosamiento del cuerpo calloso y la evolución maligna son inconsistentes con estas entidades como causa de su proceso.
La administración de diversos fármacos y tóxicos también pueden causar una lesión de la sustancia blanca cereral. Entre ellos cabe mencionar la ciclosporina y diversos citostáticos, la heroína adulterada, algunos solventes y pesticidas, y el hexaclorofeno. En este caso ninguna de estas circunstancias estaba presente.
Diversas enfermedades metabólicas pueden ocasionar leucopatía; el prototipo lo constituye la enfermedad de Canavan, une enfermedad eminentemente pediátrica cuyo sustrato anatomopatológico incluye la espongiosis. La malignidad de este caso, que además se presentó en la sexta década, hace descartable esta entidad. Además, las enfermedades mitocondriales pueden acompañarse de leucoencefalopatía, como es el caso del síndrome POLIP (polineuropatía, oftalmoplejia, leucoencefalopatía y pseudo-obstrucción intestinal). Sin embargo, todas estas enfermedades cursan de un modo mucho más indolente, por lo que son descartables. Cabe mencionar, dada la afectacón del cuerpo calloso presente en esta paciente, la enfermedad de Marchiafava-Bignami, una necrosis del cuerpo calloso en sus dos tercios anteriores que sucede en bebedores importantes. El cuadro clínico es grave, pero en esta paciente no había antecedente de ingesta etílica, y las extensas lesiones de sustancia blanca son inconsistentes con este diagnóstico.
Las infecciones pueden ser causa de leucopatía, principalmente la infección por virus JC o leucoencefalopatía multifocal adquirida. Se trata de una enfermedad de curso subagudo en la que la histopatología cerebral muestra áreas de desmielinización con presencia de oligodendrocitos distorsionados y con inclusiones virales. No ocurre, sin embargo, espongiosis. La RMN craneal muestra lesones multifocales de la sustancia blanca, con cierta preferencia por las regiones posteriores de los hemisferios cerebrales. Se trata fundamentalmente de una infección oportunista del paciente inmunosuprimido, aunque ocasionalmente afecta al inmunocompetente. En nuestros días constituye un problema en el paciente con SIDA, pues se estima que un 4% de los fallecidos con SIDA tienen esta infección. En ocasiones, diversas infecciones sistémicas dan lugar a una leucopatía, pero nunca tan grave ni tan extensa como la aquí presentada, y en cualquier caso, la biopsia no demostraría espongiosis. Tal es el caso de la neuroborreliosis o la infección por Mycoplasma pneumoniae, entre otras.
Entre la causas tumorales de leucopatía difusa se encuentran el linfoma cerebral, fundamentalmente en su forma intravascular, y la gliomatosis cerebri. El linfoma intravascular afecta principalmente al sistema nervioso y la piel. Las células linfomatosas quedan confinadas al espacio intravascular debido a la expresión de moléculas de adhesión al endotelio. La oclusión vascular resultante da lugar a infartos múltiples, mielopatía, encefalopatía aguda o neuropatía periférica. Su pronóstico es sombrío, con una supervivencia media de unos 5 meses. En este caso, sin embargo, la afectación tan marcada de la sustancia blanca, sin distribución vascular, no es consistente con esta entidad. Además, la biopsia no mostró los hallazgos característicos. Por otro lado, el linfoma cerebral responde inicialmente a los esteroides, y en esta paciente no hubo respuesta. La gliomatosis cerebri es una entidad neurológica infrecuente y de mal pronóstico, con una supervivencia media de 3-6 meses (1). Se caracteriza por una infiltación tumoral difusa que afecta a más de dos lóbulos cerebrales. El diagnóstico no es fácil (2) y su presentación clínica es variada, incluyendo principalmente el deterioro cognitivo, cambios en la personalidad, crisis epilépticas, ataxia, y déficits neurológicos focales. Como en esta paciente, la RMN cerebral muestra afectación difusa de la sustancia blanca con infiltración del cuerpo calloso; a diferencia de ella, suele haber infiltración de los ganglios de la base y captación de gadolinio en la mitad de los casos (1). La secuencia FLAIR, que suprime la señal del LCR pemitiendo así visualizar mejor las áreas periventriculares, es más sensible para la detección de las lesiones en esta entidad (3); en particular, ayuda a delinear la lesión del cuerpo calloso. Una serie reciente analiza 13 casos de esta entidad que fueron diagnosticados por biopsia cerebral (1) . En todos ellos se demostraba un grado variable de infiltración parenquimatosa cerebral por células de estirpe glial cuyo origen es aún objeto de debate. Por tanto, el cuadro clínico, la evolución rápida y maligna y los hallazgos de la neuroimagen son consistentes con los de la paciente que aquí se discute. A mi juicio, ésta constituye la sospecha clínica principal, y pienso que los clínicos la barajaban al solicitar una biopsia cerebral, pues esta entidad puede ser susceptible de tratamiento quimioterápico (4) o radioterápico (5, 6). Sin embargo, la presencia de espongiosis en la biopsia cerebral es desconcertante, pues no estaba presente en ninguno de los 13 pacientes de la serie anteriormente aludida (1).
Finalmente, las encefalopatías espongiformes y transmisibles constituyen el representante clásico de una entidad devastadora del sistema nervioso y que se caracteriza por la presencia de espongiosis en el tejido cerebral. Esta entidad, transmisible, puede ocurrir tanto en animales (scrapie, encefalopatía del visón y otras) como en el ser humano. Esta última fue descrita por Jakob y Creutzfeldt en Alemania en la década de los 1920 en varios pacientes con demencias atípicas de evolución rápida y fatal en los que el estudio anatomopatológico cerebral evidenciaba una pérdida neuronal con gliosis e importante espongiosis. El agente etiológico de esta enfermedad es el prión, una proteína (PrP) que se deposita en el tejido neural al adquirir una conformación anómala. La forma de presentación más frecuente es la esporádica, aunque existen formas familiares (5-10%) relacionadas con mutaciones en el gen de la PrP. A nivel clínico se presenta con una demencia progresiva con mioclonías y afectación de diversas estructuras del sistema nervioso (piramidal, extrapiramidal, visual) en diferentes combinaciones. Como en la paciente que aquí se discute, el inicio suele ser muy inespecífico, en forma de insomnio, depresión, pérdida de peso, astenia e hipersudoración. El EEG muestra —en algún momento de la evolución— actividad periódica, aunque ésta no es exclusiva de esta enfermedad. El estudio neurofisiológico del sueño (polisomnografía) demuestra una desestructuración del sueño, como se objetivó en esta paciente, aunque tampoco es un hallazgo específico. La RMN craneal muestra típicamente hiperintensidades en los ganglios de la base y corteza cerebral, que se detectan mejor con secuencias FLAIR. Las lesiones de la sustancia blanca no son frecuentes en esta enfermedad, pero se conoce desde hace años que la variedad conocida como panencefalopática afecta típicamente a la sustancia blanca. Además, en modelos animales se ha demostrado que existe una afectación inicial de la sustancia blanca que precede a la de la sustancia gris (7). Además, algunos estudios han encontrado una alteración clara de la ologodendroglía en esta enfermedad (8). Una prueba importante en el diagnóstico es la detección en el LCR de la proteína 14-3-3, una proteína neuronal que se encuentra elevada en un 95% de las formas esporádicas. Es chocante, por tanto, que si esta paciente tuviera una prionopatía con niveles normales. Sin embargo, existen casos descritos y es posible que dichos niveles sean normales con en los períodos precoces de la enfermedad. Finalmente, la presencia de espongiosis junto a la detección de proteína priónica (PrP) en el tejido cerebral constituyen la confirmación histológica de esta enfermedad. En esta paciente, existía espongiosis en las dos biopsias, pero la tinción para PrP en tejido neural fue negativa. Ciertamente estos hallazgos son atípicos para las prionopatías, pero el conocimiento acumulado recientemente indica que hay diversas variedades de enfermedad, determinadas por la cepa de prión y por el genotipo de un codón (el 129) del gen que codifica la proteína PrP, que cursan con diferente cuadro clínico, localización anatómica y en las que la detección de la proteína 14-3-3 no es tan frecuente. El depósito proteínico no es homogéneo, lo que puede conducir ocasionalmente a la negatividad para PrP de las biosias cerebrales. Las confusiones diagnósticas relacionadas con esta enfermedad son frecuentes, como lo demuestran las múltiples descripciones de pacientes con biopsia cerebral compatible con ECJ cuando el cuadro clínico no era sugestivo, como en el caso de la presentación ictal de esta entidad (9). El riesgo de transmisión de esta enfermedad obliga a tomar las precauciones adecuadas ante toda biopsia cerebral. La situación inversa también ocurre, en la que la sospecha inicial de ECJ se torna, por ejemplo, en una tiroiditis de Hashimoto (10), incluso con evidencia de espongiosis cerebral; la presencia de inflamación acompañante distingue las dos entidades, pues está ausente en las prionopatías.
Llegamos, por tanto, a un dilema diagnóstico en el que el cuadro de la paciente, un deterioro cognitivo progresivo y letal en 4 meses, aunque no específico, es consistente con una prionopatía o una enfermedad tumoral. La neuroimagen, con una afectación difusa de la sustancia blanca y el engrosamiento del cuerpo calloso claramente orienta más a la segunda posibilidad, en este caso una gliomatosis cerebri. Sin embargo, la biopsia demostró espongiosis, que nos llevaría de nuevo a la primera entidad.
Por tanto, existen dos posibilidades diagnósticas fundamentales en esta paciente: una enfermedad esporádica por priones con afectación preferente de la sustancia blanca, no pudiéndose descartar la gliomatosis cerebri.
ANATOMIA PATOLOGICA (Dr. J. Figols)
La autopsia , limitada a cavidad craneal, mostró un encéfalo de 1350 gr (fijado), que al examen externo mostraba circunvoluciones ensanchadas (lo que era más patente en hemisferio derecho), con surcos parcialmente borrados. Herniación uncal bilateral y prominencia amigdalar (fig. 10).

Fig. 10.
Se observaron los puntos de entrada de las biopsias esterotáxicas practicadas, una en frontal y otra en parietal, ambos en el hemisferio derecho.
A los cortes coronales se constató ampliación de los centros semiovales con aumento de la consistencia, así como del cuerpo calloso (1,3 cm de espesor) con desplazamiento de la línea media por ampliación del hemisferio derecho y «borramiento-adelgazamiento» del cortex frontal de ese lado (figs. 11 y 12).
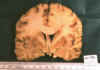
Fig. 11.
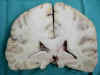
Fig. 12.
En el corte correspondiente al máximo desarrollo del tálamo y del asta de Ammon (fig. 13), aparece una neoformación abigarrada ocupante de espacio, con áreas necrótico-hemorrágicas con edema peritumoral. Esta tumoración, en cortes sucesivos se extiende hasta el corte del máximo desarrollo del pulvinar.

Fig. 13.
El estudio microscópico de la sustancia blanca, en el resto de las áreas fuera de la tumoración descrita, tanto de los centros semiovales (de forma bilateral), como del cuerpo calloso, muestra una proliferación anormal de elementos celulares alargados en su mayoría, de morfología astrocitaria (figs. 14 y 15), a veces con citoplasmas amplios, casi gemistocíticos (fig. 14) alternando con áreas de mayor densidad celular (fig. 15), que afectaba a la totalidad de la sustancia blanca cerebral, y llegando hasta la calota mesencefálica a través de la vía piramidal. Estos astrocitos no muestran rasgos de malignidad citológica y aunque en algunas zonas, como se observa en la figura 15, se observe mayor densidad, era muy difícil encontrar figuras de mitosis.
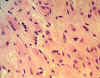
Fig. 14.
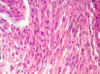
Fig. 15.
En otras zonas (fig. 16) se hallaron también morfologías astrocitarias más estrelladas y abigarradas, a veces con multinucleaciones.

Fig. 16.
Los cortes practicados a la neoformación frontal derecha, revelaron una típica estructura glioblástica, con alta densidad celular, necrosis en «empalizada» y vasos glomeruloides (fig. 17). La neoformación glioblástica y el resto de la celularidad de sustancia blanca fue positiva para GFAP (fig. 18).
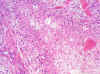
Fig. 17.
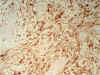
Fig. 18.
La corteza no mostró anomalías microscópicas a ningún nivel, no objetivándose gliosis ni espongiosis. La PrP fue también negativa. Por debajo de la corteza se observó una preservación relativa de las fibras en «U» subcorticales, y un área de edema (con espongiosis de la sustancia blanca que la separaba de la proliferación astrocitaria y del glioblastoma. Estas alteraciones explican el que no se hallaran dichos astrocitos anormales en las biopsias practicadas a la paciente (fig. 19).


Fig. 19.
DISCUSIÓN ANATOMOPATOLÓGICA
El estudio del cerebro mostró ausencia total de signos histológicos e inmunohistoquímicos de prionopatía como ya se ha referido, por lo que se descartó completamente dicho diagnóstico que fue sospechado en las primeras biopsias por aguja por la existencia de la gliosis y espongiosis (aunque tal lesión se manifestaba de una manera exclusiva en sustancia blanca). Ya desde el estudio macroscópico del cerebro en sus cortes coronales se observó que se trataba de una leucopatía difusa con un engrosamiento notable del cuerpo calloso, lo que se correlacionaba muy bien con las imágenes de la RMN (compárense las figuras 5 y 6, con la 11 y la 12). La neoformación frontal derecha no tiene correlato radiológico, lo que se explicaría por ser un foco de malignización que surgió y creció muy rápidamente (en un mes aproximadamente desde la última RMN). El estudio microscópico de la sustancia blanca nos reveló una gliomatosis difusa (afecta a más de 2-3 lóbulos cerebrales) circunscrita a sustancia blanca sin afectación cortical, compuesta de astrocitos en su mayoría bipolares, como es lo más frecuente en las gliomatosis, pero también multipolares y gemistocíticos en focos. La neoformación derecha ya referida, se trataba de un glioblastoma, muy verosimilmente desarrollado por transformación-desdiferenciación glioblástica de los astrocitos componentes de la gliomatosis, surgida bruscamente en un momento concreto de la evolución de la paciente, probablemente en momentos cercanos a la fecha del éxitus. El enclavamiento uncal, y amigdalar así como parcialmente de la circunvolución supracallosa, sugieren esta última apreciación que pudo además ampliarse por la aparición de necrosis y/o hemorragias bruscas en el seno del glioblastoma, con el consiguiente edema y aumento de masa.
La gliomatosis cerebri , también llamada gliomatosis difusa, es un término acuñado por Nevin en una primera publicación en 1938 (1), tema posteriormente tratado en profundidad por algunos autores (2) .Se trata de una patología de muy baja incidencia, de tal forma, que las mayores series publicadas no suelen pasar de 4 casos (3,4). Se definió en las primeras publicaciones como una proliferación celular difusa «sin masa tumoral individualizada», que afecta a varios lóbulos cerebrales (eventualmente al tronco y a la médula) y que característicamente respeta la arquitectura anatómica.
En relación a esta entidad, se ha debatido siempre si se trata de una entidad patológica en sí o bien es más bien un tipo especial de astrocitoma que infiltra difusamente. En 1995 Hechst y cols (5), demuestran diferencias en las alteraciones cromsómicas en una y otra entidad, y posteriormente en 1997, Kattar y cols (6), hallan inestabilidad de microsatélites en la gliomatosis lo que apoya también la neta separación entre la gliomatosis y el astrocitoma difuso. La OMS, en su última revisión del año 2000 ha aceptado por fin la gliomatosis dentro del capítulo de «tumores neuroepiteliales de origen incierto».
Afecta a pacientes de todas las edades y aunque las imágenes con RMN en T1 no muestran una lesión muy contrastada, este contraste se incrementa de una manera muy notable en secuencias T2 o en FLAIR.
Aunque la celularidad predominante en los casos publicados es de naturaleza astrocitaria, se han descrito algunos casos en los que las células que componen la gliomatosis son oligodendrogliales (1,7). En la mayoría de las series la densidad celular es baja, y el astrocito es de apariencia pilocítica , en algunos casos o en focos aislados, de aspecto estrellado. Naturalmente, el marcador inmunohistoquímico, al tratarse de células de naturaleza astrocitaria es la proteína fibrilar glial (PAGF) (8). No obstante el aspecto poco agresivo de la mayor parte de los casos, se han descrito áreas de transformación anaplásica ocasionalmente. De hecho, en un artículo muy reciente (9),se admite ya claramente la posibilidad de que en el seno de una gliomatosis se origine algún foco de transformación glioblástica. Esta posibilidad ha dado pie a algunos autores (10) a subdividir la gliomatosis en dos tipos: el tipo I, que correspondería a la forma clásica de celularidad difusa, y el tipo II, en el que en el seno de la proliferación difusa gliomatosa se origine un foco tumoral diferenciado, como tiene lugar en nuestro caso, que pertenecería a este segundo tipo.
BIBLIOGRAFÍA DE LA DISCUSIÓN CLÍNICA (Dr. García Moncó)
Herrlinger U, Felsberg J, Kuker W, Bornemann A, Plasswilm L, Knobbe CB, et al. Gliomatosis cerebri: molecular pathology and clinical course. Ann Neurol 2002; 52: 390-9.
Rust P, Ashkan K, Ball C, Stapleton S, Marsh H. Gliomatosis cerebri: pitfalls in diagnosis. J Clin Neurosci 2001; 8: 361-3.
Freund M, Hahnel S, Sommer C, Martmann M, Kiessling M, Tronnier V, et al. CT and MRI findings in gliomatosis cerebri: a neuroradiologic and neuropathologic review of diffuse infiltrating brain neoplasms. Eur Radiol 2001; 11: 309-16.
Louis E, Keime-Guibert F, Delattre JY, Sanson M. Dramatic response to chemotherapy in oligodendroglial gliomatosis cerebri. Neurology 2003; 60: 151.
Elshaikh MA, Stevens GH, Peereboom DM, Cohen BH, Prayson RA, Lee SY, et al. Gliomatosis cerebri: treatment results with radiotherapy alone. Cancer 2002; 95: 2027-31.
Horst E, Micke O, Romppainen ML, Pyhtinen J, Paulus W, Schafer U, et al. Radiation therapy approach in gliomatosis cerebri—case reports and literature review. Acta Oncol 2000; 39: 747-51.
Kordek R, Hainfellner JA, Liberski PP, Budka H. Deposition of the prion protein (PrP) during the evolution of experimental Creutzfeldt-Jakob disease. Acta Neuropathol (Berl) 1999; 98: 597-602.
El Hachimi KH, Chaunu MP, Brown P, Foncin JF. Modifications of oligodendroglial cells in spongiform encephalopathies. Exp Neurol 1998; 154: 23-30.
Doherty CP, Schlossmacher M, Torres N, Bromfield E, Samuels MA, Folkerth R. Hashimoto’s encephalopathy mimicking Creutzfeldt-Kacob disease: brain biopsy findings. J Neurol Neurosurg Psychiatry 2002; 73: 601-2.
Whittle IR, Will RG, Ironside JW. Brain biopsy and patients with atypical presentations of sporadic Creutzfeldt-Jacob disease. J Neurol Neurosurg Psychiatry 1997; 63: 547-8.
BIBLIOGRAFÍA DE LA DISCUSIÓN NEUROPATOLÓGICA
Nevin S. Gliomatosis cerebri. Brain 1938; 61: 170-191.
Artigas J, Cervós-Navarro J, Iglesias JR, Ebhardt G. Gliomatosis cerebri: clinical and histological findings. Clin Neuropathol, 1985; 4: 135-148.
Couch JR, Weiss SA. Gliomatosis cerebri: Report of four cases and review of the literature. Neurology 1974; 24: 504-511.
Kandler RH, Smith CM, Broome JC, Davies-Jones GA. Gliomatosis cerebri: a clinical, radiological and pathological report of four cases. Br J Neurosurg 1991; 5: 187-193.
Hecht BK, Turk-Carel C, Chatel M, et al. Chromosomes in gliomatosis cerebri. Genes Chromosomes Cancer 1995; 14: 149-153.
Kattar MM, Kupsky WJ, Shimoyama RK et al. Clonal analysis of gliomas. Hum Pathol 1997; 28: 1166-1179.
Balko MG, Blisard KS, Samaha FJ. Oligodendroglial gliomatosis cerebri. Hum Pathol 1992; 23: 706-707.
Figols J, Cruz-Sánchez F. Inmunohistoquímica de los tumores del Sistema Nervioso Central y Periférico. En Escalona-Zapata, editor. Tumores del Sistema Nervioso Central. Madrid, 1996. p: 674.
Burger PC. Gliomatosis cerebri: Molecular pathology and clinical course. Ann Neurol 2002; 52: 389.
Hoang-Xuan K, Dairou R, Gray F, Sellal F. Confrontation de la Salpêtrière. Crises d’épilepsie et hémiparesie gauche à répetition. Rev Neurol (Paris) 2002; 158: 369-376.
![]()