
Vol. 36, n.º 4, 2003
|
REVISTA
ESPAÑOLA DE
Vol. 36, n.º 4, 2003 |
F. Ignacio Aranda, Cristina Alenda, Francisca M. Peiró, Gloria Peiró
Servicio de Patología. Hospital General Universitario de Alicante. aranda_ign@gva.es
RESUMEN
Los tumores neuroendocrinos pulmonares constituyen un grupo bien definido dentro de la clasificación de la OMS de 1999. La clasificación de los tumores carcinoides en típicos y atípicos basada en criterios morfológicos permite establecer el pronóstico, con independencia del estadio. La incorporación del carcinoma neuroendocrino de célula grande, más agresivo que los tumores carcinoides atípicos, permite diferenciar estos casos del carcinoma de célula pequeña o del carcinoma de célula grande clásico. En esta revisión, se discuten las categorías diagnósticas, haciéndose referencia a los aspectos clínicos, histológicos, inmunohistoquímicos y moleculares.
Palabras clave: tumor carcinoide típico, carcinoide atípico, carcinoma neuroendocrino de célula grande, carcinoma de célula pequeña.
SUMMARY
The 1999 WHO classification establishes neuroendocrine tumors of the lung as separate entities. Carcinoid tumors, by morphological criteria, are considered typical or atypical. This differentiation permits the establishment of prognoses, regardless of their stage.
The inclusion of large-cell neuroendocrine carcinoma of the lung, which is more aggressive than atypical carcinoid, permits distinguishing these cases from small-cell carcinoma or classic large-cell carcinoma. Current diagnostic categories (including clinical, histological, ultrastructural, immunohistochemical and molecular features) are discussed in this review.
Key words: carcinoid tumor, atypical carcinoid, large cell neuroendocrine tumor, small cell lung carcinoma.
INTRODUCCIÓN
Los tumores neuroendocrinos pulmonares representan un 20% de los carcinomas pulmonares y constituyen un grupo heterogéneo de neoplasias con morfología, comportamiento y pronóstico muy diferentes (1-4). Durante años las dos únicas entidades reconocidas y discutidas de este grupo fueron el tumor carcinoide (TC) y el carcinoma de célula pequeña (CCP) (5,6). Aunque la distinción entre tumores carcinoides típicos (TCT) y atípicos (TCA) fue descrita por Engelbrech-Holm en 1944 (7), no fue hasta 1972 en que Arrigoni propone unos criterios histológicos para separar estos dos tipos, al observarse un comportamiento más agresivo de los TCA (8). En 1991 se describieron de forma pormenorizada los criterios diagnósticos del carcinoma neuroendocrino de célula grande (CNECG), propuesto como un cuarto miembro del grupo de los tumores neuroendocrinos pulmonares (1). La nueva clasificación de la OMS de 1999 ha establecido una mejor definición de los tipos histológicos, basada en datos morfológicos convencionales e inmunohistoquímicos (tabla 1), con implicaciones pronósticas y terapéuticas (9). En la presente revisión se aborda el grupo de neoplasias con morfología neuroendocrina y los carcinomas no célula pequeña con diferenciación neuroendocrina.

TUMOR CARCINOIDE TÍPICO Y ATÍPICO
Presentación clínica y características macroscópicas
Los TC se consideran neoplasias malignas de bajo grado y constituyen un 1-2% de los tumores pulmonares (2,10-12). Los hallazgos clínicos más comunes son la hemoptisis, la neumonía obstructiva, la infección pulmonar recidivante y la disnea (12,13). Ocurren con igual frecuencia en hombres y mujeres y la edad media de aparición son los 55 años. Pueden aparecer en niños y adolescentes, donde es el tumor bronquial más frecuente. Se asocian con síndromes paraneoplásicos como síndrome carcinoide, síndrome de Cushing y acromegalia. Pueden aparecer en el contexto de síndrome de MEN 1 o en formas familiares sin mutación del gen del MEN 1 (14).
Los TC pueden ser centrales o periféricos (16-40%) (2,13,14). Los centrales tienen un tamaño medio de 3 cm, y presentan componente endobronquial en forma de masa polipoide que protruye en la luz (fig. 1). Este crecimiento es más común en los carcinoides típicos que en los atípicos. La superficie de sección puede ser rojiza o amarillenta. El tumor puede infiltrar el cartílago bronquial y extenderse al parénquima pulmonar adyacente. Por el contrario, los TC periféricos tienden a ser más pequeños, 2,5 cm de media, están situados en el parénquima subpleural y a menudo no presentan relación anatómica con la pared bronquial (fig. 2). Suelen estar bien delimitados pero no encapsulados, ser múltiples y asociarse a tumorlets (11). En un 39% de los TC centrales y en un 8% de los periféricos se puede identificar calcificación intratumoral con radiología (15).
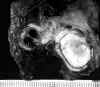
Fig. 1. Tumor carcinoide central. Masa
que ocupa la luz bronquial con dilatación de la misma, en pieza de lobectomía.
Fig. 2. Tumor carcinoide periférico.
Tumoración de límites bien definidos de localización subpleural. Resección
atípica.
Los TCA constituyen entre el 10 y el 25 % de los TC pulmonares y presentan un diámetro medio mayor (3,6 cm) que los típicos (1,2,8,11-13,16,17).
Características microscópicas
Los criterios diagnósticos de los TC pulmonares redefinidos en 1998 por Travis han sido incorporados a la última clasificación de la OMS (9) (tabla 2). Los TCT se caracterizan por presentar morfología neuroendocrina (fig. 3), menos de 2 mitosis por 2 mm2 (10 campos de gran aumento-CGA-) y ausencia de necrosis (3,9). Los TCA también presentan morfología neuroendocrina, con 2-10 mitosis por 2 mm2 o necrosis. Los TCT y TCA se caracterizan por su arquitectura organoide y morfología uniforme. Están constituidos por células moderadamente eosinófilas con núcleos de cromatina finamente granular. Los nucleolos son poco prominentes. Muestran múltiples patrones histológicos, que además pueden coexistir: trabecular (fig. 4A), glandular, acinar (fig. 4B), folicular, papilar, esclerosante, fusiforme (fig. 4C) y difuso. La celularidad puede adoptar morfología oncocítica, acinar, en anillo de sello o de célula clara. Hasta en un 60% se demuestra la formación de mucina (20), si bien este hallazgo no tiene significado pronóstico. De manera excepcional presentan cuerpos hialinos intracitoplasmáticos (fig. 4D), que corresponden a ovillos de filamentos (21), con tinción para citoqueratina positiva. El estroma suele ser fibroso y, frecuentemente presenta calcificaciones u osificación. En ocasiones se observa depósito de amiloide (17,22). Los TCA, además de un mayor número de mitosis o necrosis (fig. 5A y 5B) presentan pleomorfismo nuclear, hipercromatismo, alteración de la relación núcleo-citoplasmática, aumento de la densidad celular, y desorganización de la arquitectura (1,8,23,24). El número de mitosis y el tamaño tumoral serían factores pronósticos negativos dentro del grupo de TCA (17). Tanto en los TCT como en los TCA puede observarse invasión vascular (fig. 5C).

Se ha descrito aneuploidía en 5-32% de los TCT y en el 16-79% de los TCA. Al igual que en otros tumores endocrinos, la aneuploidía no necesariamente indica mal comportamiento biológico (25,26).

Fig. 3. Tumor carcinoide típico.
Arquitectura organoide, sin actividad mitótica significativa ni necrosis (H-E
x200).

Fig. 4. Arquitectura y celularidad en
tumor carcinoide atípico. (A) Arquitectura trabecular (H-E x400) y (B)
arquitectura acinar (H-E x200). (C) Celularidad fusiforme (H-E x200). (D)
Células con citoplasma claro, hialino (H-E x400).

Fig. 5. Tumor carcinoide típico y
atípico. (A) Figuras de mitosis y (B) focos de necrosis en tumor carcinoide
atípico. (C) Imágenes de invasión vascular en tumor carcinoide típico (H-E
x200). (D) Metástasis de tumor carcinoide atípico en ganglio linfático hiliar
(H-E x100).
Aspectos citológicos
La citología de esputo rara vez es diagnóstica. Sin embargo la PAAF permite el diagnóstico al identificarse células pequeñas u ovales con núcleos redondos o alargados, que se disponen de forma dispersa o formando pequeños agregados. La cromatina es finamente granular. Los atípicos presentan mayor grado de pleomorfismo nuclear y nucleolos por lo que pueden ser más difíciles de diferenciar de un carcinoma (27).
Ultraestructura
La principal característica ultraestructural de los TC es la presencia de numerosos gránulos densos con considerable variación en la forma y el tamaño (90-450 nm) (1,28). Los TC oncocíticos presentan un alto contenido en mitocondrias (29).
Inmunohistoquímica
Presentan positividad para citoqueratinas y los marcadores neuroendocrinos habituales como cromogranina (95%), sinaptofisina (90%) (fig. 6A), Leu7 (CD57) (75%). También son útiles otros marcadores como NCAM (neural cell adhesion molecules) (CD56) (30) o MAP-2 (microtubule-associated protein-2) (31), con sensibilidades del 100%. Los marcadores hormonales pueden confirmar la producción ectópica de ACTH, de GHRH, de VIP, o de calcitonina. Las metástasis suprarrenales de carcinoides productores de ACTH pueden asociarse a hiperplasia corticosuprarrenal (32). En ocasiones presentan un patrón de positividad para citoqueratina en gota (fig. 6B) similar al observado en los tumores de células de Merckel o en los adenomas hipofisarios somatotropos pobres en gránulos. Los marcadores de proliferación celular como Ki-67 demuestran que los TCT presentan actividad proliferativa baja (en general, menos del 10%) (fig. 6C). El anticuerpo anti-proteína mitocondrial demuestra el alto contenido de mitocondrias en casos con cambio oncocítico (fig. 6D). Los TCA presentan mayor actividad proliferativa (33) e índice de apoptosis (34). El TTF-1 (thyroid transcription factor 1) permite diferenciar un TC primario de metástasis pulmonares por tumores neuroendocrinos de diversos orígenes (35).

Fig. 6. Inmunohistoquímica en tumor
carcinoide. (A) Tumor carcinoide típico con arquitectura trabecular.
Inmunotinción con sinaptofisina (EBP x400). (B) Patrón "en gota",
paranuclear con citoqueratina en tumor carcinoide típico (AE1/AE3 EBP x400).
(C) Tinción para Ki-67 en tumor carcinoide típico con actividad proliferativa
baja (EBP x400). (D) Inmunotinción mitocondrial en tumor carcinoide con cambio
oncocítico (antimitocondrial EBP x400).
Patología molecular
Los estudios iniciales con p53 parecían demostrar que la sobreexpresión inmunohistoquímica de esta proteína estaba restringida a los TCA (45-64%) (36-38). Sin embargo, estudios posteriores sólo encuentran casos aislados con positividad focal en los TC (32,39-41). Los estudios moleculares no demuestran mutación de los exones 5, 7 y 8 del gen de la p53 en los TCT ni en los TCA (37). Esto sugeriría que las alteraciones en este gen no juegan ningún papel, al menos en la mayoría de los casos de TC.
En relación con el gen del retinoblastoma (RB), estudios inmunohistoquímicos no demuestran ausencia de expresión de esta proteína en los TCT. En el TCA los resultados son variables, observándose pérdida de RB en el 20%. Por contra, la mayor parte de los casos de CCP y de CNECG son negativos para la proteína de RB, indicativo de pérdida de función del gen (33,42-44).
La proteína bcl-2, al contrario de lo que ocurre en el CCP y en el CNECG, parece ser negativa en la mayoría de los casos de TCT (38,39,45). La mayor parte de los TCT y TCA sobreexpresan Bax, lo que indica una relación bcl-2/bax menor de 1, lo contrario de lo que ocurre en el CNECG y en el CCP (expresión alta de bcl-2 y baja de Bax) (39). Los TCT no presentan aumento de la actividad telomerasa, que sí puede observarse en los TCA, en los CNECG y en los CCP (46,47).
Diversos estudios en TC han demostrado pérdidas de heterozigosidad (LOH) en diversas regiones cromosómicas como pérdidas en 11q (13-55%), en 3p (0-73%) y con menos frecuencia, en 5q, 9p y 17 p (48-52).
Tratamiento y pronóstico
El tratamiento de elección es la cirugía, mediante lobectomía con disección ganglionar, siempre que sea posible (53,54). Un 10-28% de los TCT presentan metástasis en el momento del diagnóstico (13,55,56) (fig. 5D). La supervivencia a los 10 años se acerca al 90%, incluso en casos con metástasis (3,13,55,57). Entre un 25-100% de los TCA pueden presentar metástasis en el momento del diagnóstico (55-57). En los casos con metástasis sistémicas puede realizarse tratamiento adyuvante con estreptotozina, 5-fluorouracilo, doxorrubicina, cisplatino o etopósido (58). En casos en que se demuestra receptores de somatostatina pueden ser tratados con análogos (59). Los TCT, con independencia del estadio, presentan buen pronóstico con una supervivencia a los 10 años de casi el 90%. En el caso de los TCA la supervivencia a los 10 años se reduce a un 25-69% (1,3,17,24,55,57).
CARCINOMA NEUROENDOCRINO DE CÉLULA GRANDE
Los CNECG constituyen un 2-3% de las neoplasias malignas pulmonares. Se caracterizan por presentar con técnicas convencionales un patrón organoide, trabecular (fig. 7A), rosetoide o en empalizada que sugiere diferenciación neuroendocrina, confirmada con inmunohistoquímica o microscopía electrónica (1,3,17,24,55,57), y por mostrar más de 10 mitosis por 2 mm2 (media 70 mitosis por 2 mm2). Son tumores grandes, con un tamaño medio de 3 cm (rango 1,3-10 cm), que aparecen en pacientes fumadores con una edad media de 64 años. Pueden ser centrales o periféricos y presentan a la sección áreas de necrosis y hemorragia (1,2). Con frecuencia existen metástasis en ganglios linfáticos. Antes de la descripción de este tipo tumoral, los casos se clasificaban como carcinomas mal diferenciados de célula grande o TCA. En una revisión de 766 resecciones quirúrgicas de cáncer de pulmón, Jiang y cols. identifican 22 casos (2,7%) de CNECG, la mayoría previamente clasificados como carcinoma escamoso (9 casos), adenocarcinoma (8 casos) o adenoescamoso (1 caso) (60).

Fig. 7. Carcinoma neuroendocrino de
célula grande. (A) Células poligonales, atípicas con arquitectura trabecular
(H-E x200). (B) Carcinoma neuroendocrino de célula grande combinado con
adenocarcinoma (H-E x200) (C) Inmunotinción con cromogranina A, positiva en el
componente neuroendocrino y negativa en las células de adenocarcinoma (EBC
x200). (D) Positividad para TTF1 en el componente neuroendocrino y en el
adenocarcinoma (EBP x200).
Características microscópicas
Presentan arquitectura de tumor endocrino y están constituidos por células grandes, poligonales, que muestran núcleos con nucleolos prominentes y frecuentes figuras de mitosis (1,2,3,60). La inmunohistoquímica demuestra positividad para marcadores neuroendocrinos como cromogranina, sinaptofisina, Leu-7 o bombesina. También puede observarse positividad para ACTH, péptido relacionado con el gen de la calcitonina (CGRP), calcitonina, alfa-subunidad de hormona coriogonadotropa, serotonina, polipéptido pancreático, somatostatina y péptido liberador de gastrina (pGRP) (60). Con Ki-67, más del 90% de los casos presentan actividad proliferativa superior al 50% (33). La diferenciación divergente no es fenómeno raro (20,61) (figs. 7B, 7C, 7D). Hasta un 60% muestran secreción de mucina. Si presentan más de 5 células productoras de mucina por 10 campos de gran aumento, el tumor puede clasificarse como combinado con componente de adenocarcinoma (20). Con menos frecuencia puede observarse diferenciación escamosa. Los casos combinados con CCP se clasifican dentro de este tipo (CCP combinado).
Los CNECG son aneuploides en el 75% de los casos, cifras similares a las observadas en el CCP y mayores que en el TCA (1).
Ultraestructura
Es característica la presencia de gránulos pero, a diferencia de los TC, son más escasos y de distribución irregular (1,2). La microscopía electrónica puede demostrar signos de diferenciación divergente como formación de microvillis, vesículas de moco y tonofilamentos (20).
Patología molecular
La mayoría presentan positividad para p53, lo que les hace más cercanos al CCP (33,37,39). Al igual que el CCP y al contrario de los TC, son negativos en su mayoría para la proteína RB (33). En relación con bcl-2 tienen un comportamiento similar al CCP, con positividad para esta proteína, al contrario de lo que ocurre con el TC (45), con balance bcl-2/Bax mayor de 1 (39). Recientemente se ha demostrado sobreexpresión de c-kit en el 55% de los CNECG (62). También presentan un aumento de la actividad telomerasa (46), que se asocia con alteración de bcl-2, p53, sobreexpresión de c-kit y pérdida de RB (47).
Tratamiento y pronóstico
El tratamiento de elección es la resección con disección ganglionar completa (63), con desiguales resultados, aunque la mayoría recidiva en 6 meses (64). La quimioterapia adyuvante con cisplatino, carboplatino o ciclofosfamida puede aumentar la supervivencia en estadios iniciales (65). La supervivencia al año es del 58%, a los 5 del 27% y a los 10 del 9% (3), pronóstico significativamente peor que el observado en carcinomas no célula pequeña (60).
CARCINOMA DE CÉLULA PEQUEÑA (CCP)
Constituye un 15-25% de todos los casos de cáncer pulmonar (2,66). Se presenta habitualmente en varones, casi siempre fumadores, con edad media de 60-65 años. Generalmente se localizan en situación central (70% masa perihiliar, y metástasis en ganglios linfáticos hiliares), si bien, en ocasiones pueden ser subpleurales. Los pacientes con síndrome de Pancoast tienen tumor apical. El tamaño tumoral oscila entre 0,5-9 cm (media de 3 cm) y con frecuencia presentan necrosis, raramente cavitada.
Características microscópicas
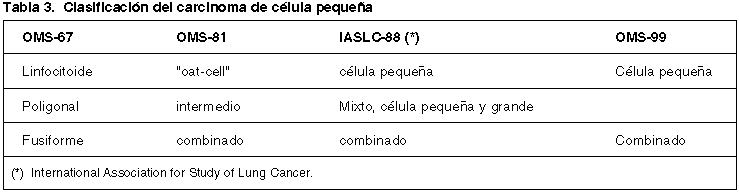
Están constituidos por células pequeñas que se disponen con patrón sólido, en nidos, trabéculas, o formando pseudorosetas. Los núcleos tienden al moldeamiento, sin nucleolo, con frecuentes fenómenos de rotura (fig. 8A) e impregnación cromatínica de vasos (fenómeno de Azzopardi). Las figuras de mitosis son numerosas (media de 80 por 2 mm2). La invasión vascular y linfática es muy común. Su subclasificación (tabla 3) tiene un bajo nivel de concordancia (67). Por ello, la nueva clasificación de la OMS incluye como única variante el tipo combinado (9). Esta variante constituye un 28% de los casos, siendo la combinación mas frecuente con carcinoma de célula grande, habitualmente de morfología neuroendocrina, seguido de adenocarcinoma y carcinoma escamoso (66).
Un 5% de los CCP pueden presentar dificultades diagnósticas, pudiendo ser clasificados como CNECG o TCA (68). El tamaño celular y la morfología del CCP y del CNECG constituyen un espectro continuo (69), a lo que se añaden problemas relacionados con el tamaño de la muestra y el artefacto de las biopsias endoscópicas. El disponer de secciones finas y bien teñidas es esencial para evaluar las diferencias (tabla 4) (66).

Inmunohistoquímica
Presentan positividad para citoqueratinas de bajo peso molecular (100%) (fig. 8B), EMA (95-100%) y CEA (55-95%). También se observa positividad de marcadores neuroendocrinos como la cromogranina A (47-60%), la sinaptofisina (5-50%) y el Leu-7 (17-40%) (66,70). En un 15% los tres marcadores neuroendocrinos pueden ser negativos (66). Un 95% son positivos para CD56, lo que puede ser de utilidad en el diagnostico diferencial con otras variedades de tumor pulmonar (71). En un 15% puede existir positividad para ACTH.

Fig. 8. Carcinoma de célula pequeña.
(A) Artefacto de estiramiento y rotura nuclear (H-E x400). (B) Positividad
citoplasmática con citoqueratina (AE1/AE3 EBP x400) y (C) para proteína bcl-2
(EBP x400). (D) Actividad alta, con positividad nuclear en la mayoría de los
núcleos para Ki-67 (EBC x400).
Ultraestructura
Como el resto de los tumores neuroendocrinos la característica más relevante es la presencia de gránulos neurosecretores, que pueden ser escasos y con un tamaño de 100 nm. El resto de las organelas están poco desarrolladas (2).
Patología molecular
La proteína bcl-2 es positiva en 50-100% de los casos (38,39,45,72,73) (fig. 8C), con balance bcl-2/Bax mayor de 1, al igual que los carcinomas neuroendocrinos de célula grande y contrariamente a lo que ocurre en los TC (39). Tras el tratamiento, la expresión de bcl-2 parece incrementarse detectándose una tinción más difusa e intensa (39). La inmunotinción con Bax parece no modificarse (39). En un 10% existe amplificación de N-myc (74), lo que se asocia a una peor respuesta a la quimioterapia (75). La mayor parte presenta positividad para la p53 (38) y los exones 5, 7 u 8 están frecuentemente mutados (37). Con Ki-67 la actividad proliferativa es mayor del 50% (fig. 8D).
La citogenética demuestra que las alteraciones más frecuentes se localizan en el cromosoma 3p (48). Las alteraciones de microsatélites en esta región se observan en la totalidad de los carcinomas de célula pequeña y en casi el 60% de los carcinomas neuroendocrinos de célula grande (48). Además se ha descrito la pérdida de DMBT1 (10q25.326.1), posible gen supresor tumoral implicado en procesos de diferenciación celular (76).
Tratamiento y pronóstico
En el momento del diagnóstico, sólo en un tercio de los pacientes la enfermedad está limitada a la cavidad torácica, y el resto tienen enfermedad diseminada (77). La cirugía no está indicada y el tratamiento inicial debe realizarse con quimioterapia, habitualmente con combinaciones de ciclofosfamida, doxorrubicina y vincristina (CAV); ciclofosfamida, doxorrubicina y etopósido; o cisplatino y etopósido (77). Las tasas de respuesta se sitúan entre el 65 y el 85%, con un 10-50% de respuestas completas. Cuando aparecen las recidivas, se han ensayado diferentes combinaciones que incluyen taxanos (paclitaxel, docetaxel), carboplatino, gemcitabina y topotecan (inhibidor de la toposomerasa I), entre otros, con respuestas de corta duración. A pesar de la respuesta, la supervivencia a los 2 años es del 40%, con una media de 15 a 20 meses.
CARCINOMAS NO CÉLULA PEQUEÑA CON DIFERENCIACIÓN NEUROENDOCRINA
Entre un 10 y un 35% de los carcinomas no célula pequeña pueden presentar diferenciación neuroendocrina cuando se utiliza inmunohistoquímica o microscopía electrónica (1,10,78,79). Esta prevalencia podría aumentar utilizando métodos de detección más sensibles (80). La positividad para bcl-2 en el carcinoma escamoso y en el adenocarcinoma se asocia a la presencia de marcadores neuroendocrinos (45). Scheleusener y cols. encuentran diferenciación neuroendocrina en el 41% de los carcinomas escamosos, en el 35% de los adenocarcinomas y en el 33% de los carcinomas indiferenciados de célula grande (81). Se ha sugerido que la diferenciación neuroendocrina, cuando se observa en más del 5% de las células, tiene un significado pronóstico negativo, pero, por otra parte, condiciona un aumento de la susceptibilidad de estos tumores a la quimioterapia (82,83), salvo en el caso de los carcinomas escamosos (83).
Dentro de los carcinomas de célula grande, han sido descritas cuatro categorías: carcinoma de célula grande clásico, carcinoma neuroendocrino de célula grande-CNECG- (ya comentado), carcinoma de célula grande sin morfología neuroendocrina pero con marcadores neuroendocrinos positivos y carcinoma de célula grande con morfología neuroendocrina y marcadores negativos (84). Algunos estudios parecen demostrar que el comportamiento biológico de los carcinomas de célula grande con morfología neuroendocrina o con marcadores neuroendocrinos sería peor que el carcinoma de célula grande "clásico" y similar al CNECG (84), y que podrían beneficiarse del tratamiento con quimioterapia adyuvante (85). Estos estudios preliminares sugieren que la diferenciación neuroendocrina tiene valor predictivo de respuesta a la quimioterapia, al menos en estadios iniciales.
BIBLIOGRAFÍA
Travis WD, Linnoila RI, Tsokos MG, Hitchcock CL, Cutler GB Jr, Nieman L, Chrousos G, Pass H, Doppman J. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometry study of 35 cases. Am J Surg Pathol 1991; 15: 529-53.
Vuitch F, Sekido Y, Fong K, Mackay B, Minna JD, Gazdar AF. Neuroendocrine tumors of the lung. Pathology and molecular biology. Chest Surg Clin N Am 1997; 7: 21-47.
Travis WD, Rush W, Flieder DB, Falk R, Fleming MV, Gal AA, Koss MN. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol 1998; 22: 934-44.
Younossian AB, Brundler MA, Totsch M. Feasibility of the new WHO classification of pulmonary neuroendocrine tumours. Swiss Med Wkly 2002; 132: 535-40.
Bensch KG, Corrin B, Pariente R, Spencer H. Oat-cell carcinoma of the lung. Its origin and relationship to bronchial carcinoid. Cancer 1968; 22: 1163-72.
Fisher ER, Palekar A, Paulson JD. Comparative histopathologic, histochemical, electron microscopic and tissue culture studies of bronchial carcinoids and oat cell carcinomas of lung. Am J Clin Pathol 1978; 69: 165-72.
Engelbrech-Holm J. Benign bronchial adenomas. Acta Chir Scan 1944; 90: 383-409.
Arrigoni MG, Woolner LB, Bernatz PE. Atypical carcinoid tumors of the lung. J Thorac Cardiovasc Surg 1972; 64: 413-421.
Travis WD, Colby T, Corrin B, Shimosato Y, Brambilla E. Histological typing of lung and pleural tumours. World Health Organization International Histological Classification of Tumours. 3rd ed., Berlin: Spriger-Verlag; 1999.
Gosney JR. Endocrine pathology of the lung. En: Antony PP, McSween RNM, editors. Recent Advances in Hispathology, n.º 16. Edinburg: Churchill-Livinstone; 1994.
Colby TV, Koss MN, Travis WD, editors. Tumors of the lower respiratory tract. Atlas of Tumor Pathology, third series, Washington DC: AFIP; 1995.
Hage R, de la Riviere AB, Seldenrijk CA, van den Bosch JM. Update in pulmonary carcinoid tumors: a review article. Ann Surg Oncol 2003; 10: 697-704.
Fink G, Krelbaum T, Yellin A, Bendayan D, Saute M, Glazer M, Kramer MR. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest 2001; 119: 1647-51.
Oliveira AM, Tazelaar HD, Wentzlaff KA, Kosugi NS, Hai N, Benson A, Miller DL, Yang P. Familial pulmonary carcinoid tumors. Cancer 2001; 91: 2104-9.
Zwiebel BR, Austin JH, Grimes MM. Bronchial carcinoid tumors: assessment with CT of location and intratumoral calcification in 31 patients. Radiology 1991; 179: 483-6.
Gould VE, Linnoila RI, Memoli VA, Warren WH. Neuroendocrine components of the bronchopulmonary tract: hyperplasias, displasias, and neoplasms. Lab Invest 1983; 49: 519-37.
Beasley MB, Thunnissen FB, Brambilla E, Hasleton P, Steele R, Hammar SP, Colby TV, Sheppard M, Shimosato Y, Koss MN, Falk R, Travis WD. Pulmonary atypical carcinoid: predictors of survival in 106 cases. Hum Pathol 2000; 31: 1255-65.
Sklar JL, Churg A, Bensch KG. Oncocytic carcinoid tumor of the lung. Am J Surg Pathol 1980; 4: 287-92.
Gaffey MJ, Mills SE, Frierson HF Jr, Askin FB, Maygarden SJ.Pulmonary clear cell carcinoid tumor: another entity in the differential diagnosis of pulmonary clear cell neoplasia. Am J Surg Pathol 1998; 22: 1020-5.
Brambilla E, Lantuejoul S, Sturm N. Divergent differentiation in neuroendocrine lung tumors. Semin Diagn Pathol 2000; 17: 138-48.
Alvarez-Fernandez E. Intracytoplasmic fibrillary inclusions in bronchial carcinoid. Cancer 1980; 46: 144-51.
Abe Y, Utsunomiya H, Tsutsumi Y. Atypical carcinoid tumor of the lung with amyloid stroma. Acta Pathol Jpn 1992; 42: 286-92.
Paladugu RR, Benfield JR, Pak PR, Ross RK, Teplitz RL. Bronchopulmonary Kulchitzky cell carcinomas. A new classifiaction scheme for typical and atypical carcinoids. Cancer 1985; 55: 1303-11.
Grote TH, Macom WR, Davis B, Greco FA, Johnson DH. Atypical carcinoid of the lung. A distinct clinicopathologic entity. Chest 1988; 93: 370-375.
Jones DJ, Hasleton PS, Moore M. DNA ploidy in bronchopulmonary carcinoid tumors. Thorax 1988; 43: 195-9.
el-Naggar AK, Ballance W, Karim FW, Ordonez NG, McLemore D, Giacco GG, Batsakis JG. Typical and atypical bronchopulmonary carcinoids. A clinicopathologic and flow cytometric study. Am J Clin Pathol 1991; 95: 828-34.
Nicholson SA, Ryan MR. A review of cytologic findings in neuroendocrine carcinomas including carcinoid tumors with histologic correlation. Cancer 2000; 90: 148-61.
Carter D, Yesner R. Carcinomas of the lung with neuroendocrine differentiation. Semin Diagn Pathol 1985; 2: 235-54.
Scharifker D, Marchevsky A. Oncocytic carcinoid of lung: an ultrastructural analysis. Cancer 1981; 47: 530-2.
Lantuejoul S, Moro D, Michalides RJ, Brambilla C, Brambilla E. Neural cell adhesion molecules (NCAM) and NCAM-PSA expression in neuroendocrine lung tumors. Am J Surg Pathol 1998; 22: 1267-76.
Liu Y, Saad RS, Shen SS, Silverman JF. Diagnostic value of microtubule-associated protein-2 (MAP-2) for neuroendocrine neoplasms. Adv Anat Pathol 2003; 10: 101-6.
Aranda FI. Metástasis suprarrenales por tumor carcinoide atípico bronquial productor de ACTH. Rev Esp Patol 1996; 29: 81-3.
Rusch VW, Klimstra DS, Venkatraman ES. Molecular markers help characterize neuroendocrine lung tumors. Ann Thorac Surg 1996; 62: 798-810.
Laitinen KL, Soini Y, Mattila J, Paakko P. Atypical bronchopulmonary carcinoids show a tendency toward increased apoptotic and proliferative activity. Cancer 2000; 88: 1590-8.
Oliveira AM, Tazelaar HD, Myers JL, Erickson LA, Lloyd RV. Thyroid transcription factor-1 distinguishes metastatic pulmonary from well-differentiated neuroendocrine tumors of other sites. Am J Surg Pathol 2001; 25: 815-9.
Roncalli M, Doglioni C, Springall DR, Papotti M, Pagani A, Polak JM, Ibrahim NB, Coggi G, Viale G. Abnormal p53 expression in lung neuroendocrine tumors. Diagnostic and prognostic implications. Diagn Mol Pathol 1992; 1: 129-35.
Przygodzki RM, Finkelstein SD, Langer JC, Swalsky PA, Fishback N, Bakker A, Guinee DG, Koss M, Travis WD. Analysis of p53, k-ras, and C-raf-1 in pulmonary neuroendocrine tumors. Correlation with histological subtype and clinical outcome. Am J Pathol 1996; 148: 1531-41.
Coppola D, Clarke M, Landreneau R, Weyant RJ, Cooper D, Yousem SA. Bcl-2, p53, and cd44v6 isoform expression in neuroendocrine tumors of the lung. Mod Pathol 1996; 9: 484-90.
Brambilla E, Negoescu A, Gazzeri S, Lantuejoul S, Moro D, Brambilla S, Coll J-L. Apoptosis-related factors p53, bcls, and bax in neuroendocrine lung tumors. Am J Pathol 1996; 149: 1941-2.
Couce ME, Bautista D, Costa J, Carter D. Analysis of K-ras, N-ras, H-ras, and p53 in lung neuroendocrine neoplasms. Diagn Mol Pathol 1999; 8: 71-9.
Sampietro G, Tomasic G, Collini P, Biganzoli E, Boracchi P, Bidoli P, Pilotti S. Gene product immunophenotyping of neuroendocrine lung tumors. No linking evidence between carcinoids and small-cell lung carcinomas suggested by multivariate statistical analysis. Appl Immunohistochem Mol Morphol 2000; 8: 49-56.
Barbareschi M, Girlando S, Mauri FA, Arrigoni G, Laurino L, Dalla Palma P, Doglioni C. Tumor suppressor gene products, proliferation, and differentiation markers in lung neuroendocrine neoplasms. J Pathol 1992; 166: 343-50.
Cagle PT, El-Naggar AK, Xu H-J, Hu S-X, Benedict WF. Differential retinoblastoma protein expression in neuroendocrine tumors of the lung. Potential diagnostic implications. Am J Pathol 1997; 150: 393-400.
Beasley MB, Lantuejoul S, Abbondanzo S, Chu WS, Hasleton PS, Travis WD, Brambilla E. The P16/cyclin D1/Rb pathway in neuroendocrine tumors of the lung. Hum Pathol 2003; 34: 136-42.
Jiang S-X, Kameya T, Sato Y, Yanase N, Yoshimura H, Kodama T. Bcl-2 protein expression in lung cancer and close correlation with neuroendocrine differentiation. Am J Pathol 1996; 148: 837-46.
Gomez-Roman JJ, Romero AF, Castro LS, Nieto EH, Fernandez-Luna JL, Val-Bernal JF. Telomerase activity in pulmonary neuroendocrine tumors: correlation with histologic subtype (MS-0060). Am J Surg Pathol 2000; 24: 417-21.
Zaffaroni N, De Polo D, Villa R, Della Porta C, Collini P, Fabbri A, Pilotti S, Daidone MG. Differential expression of telomerase activity in neuroendocrine lung tumours: correlation with gene product immunophenotyping. J Pathol 2003; 201: 127-33.
Hurr K, Kemp B, Silver SA, El-Naggar AK. Microsatellite alteration at chromosome 3p loci in neuroendocrine lung tumors. Histogenetic and clinical relevance. Am J Pathol 1996; 149: 613-20.
Onuki N, Wistuba II, Travis WD,Virmani AK, Yashima K, Brambilla E, Hasleton P Gazdar AF. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer 1999; 85: 600-7.
Payá A, Ortega E, Aranda FI, Seguí J, Niveiro M. Genetic analysis of 18 carcinoid tumors. Virchows Arch 2001; 439: 356A.
Petzmann S, Ullmann R, Klemen H, Renner H, Popper HH. Loss of heterozygosity on chromosome arm 11q in lung carcinoids. Hum Pathol 2001; 32: 333-8.
Ullmann R, Petzmann S, Klemen H, Fraire AE, Hasleton P, Popper HH. The position of pulmonary carcinoids within the spectrum of neuroendocrine tumors of the lung and other tissues. Genes Chromosomes Cancer 2002; 34: 78-85.
Cooper WA, Thourani VH, Gal AA, Lee RB, Mansour KA, Miller JI. The surgical spectrum of pulmonary neuroendocrine neoplasms. Chest 2001; 119: 14-8.
Galbis JM, Mafé JJ, Baschwitz B, Ortega E, Aranda FI, Rodriguez-Paniagua JM. Tratamiento quirúrgico de los tumores neuroendocrinos de localización broncopulmonar. Cir Esp 2003; 74: 201-5.
Garcia-Yuste M, Matilla JM, Alvarez-Gago T, Duque JL, Heras F, Cerezal LJ, Ramos G. Prognostic factors in neuroendocrine lung tumors: a Spanish Multicenter Study. Spanish Multicenter Study of Neuroendocrine Tumors of the Lung of the Spanish Society of Pneumonology and Thoracic Surgery (EMETNE-SEPAR). Ann Thorac Surg 2000; 70: 258-63.
Huang Q, Muzitansky A, Mark EJ. Pulmonary neuroendocrine carcinomas. A review of 234 cases and a statistical analysis of 50 cases treated at one institution using a simple clinicopathologic classification. Arch Pathol Lab Med 2002; 126: 545-53.
Thomas CF Jr, Tazelaar HD, Jett JR. Typical and atypical pulmonary carcinoids: outcome in patients presenting with regional lymph node involvement. Chest 2001; 119: 1143-50.
Granberg D, Eriksson B, Wilander E, Grimfjard P, Fjallskog ML, Oberg K, Skogseid B. Experience in treatment of metastatic pulmonary carcinoid tumors. Ann Oncol 2001; 12: 1383-91.
Filosso PL, Ruffini E, Oliaro A, Papalia E, Donati G, Rena O. Long-term survival of atypical bronchial carcinoids with liver metastases, treated with octreotide. Eur J Cardiothorac Surg 2002; 21: 913-7.
Jiang S-X, Kameya T, Shoji M, Dobashi Y, Shinada J, Yoshimura H. Large cell neuroendocrine carcinoma of the lung. A Histologic and immunohistochemical study of 22 cases. Am J Surg Pathol 1998; 22: 526-37.
Ruffini E, Rena O, Oliaro A, Filosso PL, Bongiovanni M, Arslanian A, Papalia E, Maggi G. Lung tumors with mixed histologic pattern. Clinico-pathologic characteristics and prognostic significance. Eur J Cardiothorac Surg 2002; 22: 701-7.
Araki K, Ishii G, Yokose T, Nagai K, Funai K, Kodama K, Nishiwaki Y, Ochiai A. Frequent overexpression of the c-kit protein in large cell neuroendocrine carcinoma of the lung. Lung Cancer 2003; 40: 173-80.
Zacharias J, Nicholson AG, Ladas GP, Goldstraw P.Large cell neuroendocrine carcinoma and large cell carcinomas with neuroendocrine morphology of the lung: prognosis after complete resection and systematic nodal dissection. Ann Thorac Surg 2003; 75: 348-52.
Mazieres J, Daste G, Molinier L, Berjaud J, Dahan M, Delsol M, Carles P, Didier A, Bachaud JM. Large cell neuroendocrine carcinoma of the lung: pathological study and clinical outcome of 18 resected cases. Lung Cancer 2002; 37: 287-92.
Iyoda A, Hiroshima K, Toyozaki T, Haga Y, Baba M, Fujisawa T, Ohwada H. Adjuvant chemotherapy for large cell carcinoma with neuroendocrine features. Cancer 2001; 92: 1108-12.
Nicholson SA, Beasley MB, Brambilla E, Hasleton PS, Colby TV, Sheppard MN, Falk R, Travis WD. Small cell lung carcinoma (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol 2002; 26: 1184-97.
Burnett RA, Howatson SR, Lang S, Lee FD, Lessells AM, McLaren KM, Nairn ER, Ogston S, Robertson AJ, Simpson JG, Smith GD, Tavadia HB, Walker F. Observer variability in histopathological reporting of non-small cell lung carcinoma on bronchial biopsy specimens. J Clin Pathol 1996; 49: 130-3.
Travis WD, Gal AA, Colby TV, Klimstra DS, Falk R, Koss MN. Reproducibility of neuroendocrine lung tumor classification. Hum Pathol 1998; 29: 272-9.
Marchevsky AM, Gal AA, Shah S, Koss MN. Morphometry confirms the presence of considerable nuclear size overlap between «small cells» and «large cells» in high-grade pulmonary neuroendocrine neoplasms. Am J Clin Pathol 2001; 116: 466-72.
Guinee DG Jr, Fishback NF, Koss MN, Abbondanzo SL, Travis WD. The spectrum of immunohistochemcial staining of small-cell lung carcinoma in specimens from transbronchial and open-lung biopsies. Am J Clin Pathol 1994; 102: 406-14.
Kaufmann O, Georgi T, Dietel M. Utility of 123C3 monoclonal antibody against CD56 (NCAM) for the diagnosis of small cell carcinomas on paraffin sections. Hum Pathol 1997: 28: 1373-8.
Alkan S, Diaz J, Baekey P. bcl-2 is commonly expressed in small cell carcinoma of lung. Am J Clin Pathol 1994; 102: 526A.
Ben-Ezra JM, Kornstein MJ, Grimes MM, Krystal G. Small cell carcinomas of the lung express the bcl2 protein. Am J Pathol 1994; 145: 1036-40.
Gazdar AF. Molecular markers for diagnosis and prognosis of lung cancer. Cancer 1992; 69: 1592-9.
Funa K, Steinholtz L, Nou E, Bergh J. Increased expression of N-myc in human small cell lung cancer biopsies predicts lack of response to chemotherapy and poor prognosis. Am J Clin Pathol 1987; 88: 216-20.
Wu W, Kemp BL, Proctor ML, Gazdar AF, Minna JD, Hong WK, Mao L. Expression of DMBT1, a candidate tumor suppressor gene, is frequently lost in lung cancer. Cancer Res 1999; 59: 1846-51.
Kelly K. New chemotherapy agents for small cell lung cancer. Chest 2000; 117: S156-62.
Linnoila RI, Mulshine JL, Steinberg SM, Funa K, Matthews MJ, Cotelingam JD, Gazdar AF. Neuroendocrine differentiation in endocrine and nonendocrine lung carcinomas. Am J Clin Pathol 1988; 90: 641-52.
Carey FA, Save VE. Neuroendocrine differentiation in lung cancer. J Pathol 1997; 182: 9-10.
Fresvig A, Qvigstad G, Halvorsen TB, Falkmer S, Waldum HL. Neuroendocrine differentiation in bronchial carcinomas of classic squamous-cell type: an immunohistochemical study of 29 cases applying the tyramide signal amplification technique. Appl Immunohistochem Mol Morphol 2001; 9: 9-13.
Schleusener JT, Tazelaar HD, Jung SH, Cha SS, Cera PJ, Myers JL, Creagan ET, Goldberg RM, Marschke RF Jr. Neuroendocrine differentiation is an independent prognostic factor in chemotherapy-treated non small cell lung carcinoma. Cancer 1996; 77: 1284-91.
Hiroshima K, Iyoda A, Shibuya K, Toyozaki T, Haga Y, Fujisawa T, Ohwada H. Prognostic significance of neuroendocrine differentiation in adenocarcinoma of the lung. Ann Thorac Surg 2002; 73: 1732-5.
Pelosi G, Pasini F, Sonzogni A, Maffini F, Maisonneuve P, Iannucci A, Terzi A, De Manzoni G, Bresaola E, Viale G.Prognostic implications of neuroendocrine differentiation and hormone production in patients with Stage I nonsmall cell lung carcinoma. Cancer 2003; 97: 2487-97.
Iyoda A, Hiroshima K, Toyozaki T, Haga Y, Fujisawa T, Ohwada H. Clinical characterization of pulmonary large cell neuroendocrine carcinoma and large cell carcinoma with neuroendocrine morphology. Cancer 2001; 91: 1992-2000.
Iyoda A, Hiroshima K, Baba M, Saitoh Y, Ohwada H, Fujisawa T. Pulmonary large cell carcinomas with neuroendocrine features are high-grade neuroendocrine tumors. Ann Thorac Surg 2002; 73: 1049-54.
![]()