
Vol. 36, n.º 4, 2003
|
REVISTA
ESPAÑOLA DE
Vol. 36, n.º 4, 2003 |
Jesús Fernando González-Palacios Martínez
Hospital Ramón y Cajal. Madrid. jglezpalacios.hrc@salud.madrid.org
Los tumores endocrinos pancreáticos (TEP) constituyen para el patólogo un importante grupo de neoplasias ya que aunque son poco frecuentes, su estudio histológico rutinario puede no ser suficiente para predecir su evolución. En la actualidad se acepta que se originan de células epiteliales multipotenciales situadas en los dúctulos pancreáticos, emparentadas con las que dan lugar al primordio pancreático que, como es bien conocido, tiene un origen común para sus dos componentes exo y endocrino (1). Así, son impropios los términos de tumores de los islotes, insulares, etc. Son neoplasias relativamente raras con una incidencia anual de menos de 1 por 100.000 personas y representan aproximadamente el 15% de las neoplasias pancreáticas (2). El aumento en la sofisticación de los métodos de laboratorio ha permitido una caracterización detallada de las endocrinopatías potencialmente asociadas a estos tumores, incluyendo el síndrome de neoplasias endocrinas múltiples tipo 1 (MEN 1, S. de Wermer) y estados hiperhormonales relacionados con la producción de neuropéptidos por las células neoplásicas, que son descritas en numerosas publicaciones (1-3) (tabla 1). Existen también excelentes revisiones referentes a la patología de los TEP, recomendándose especialmente la de E Solcia, E Capella y G Kloppel (4). Aunque los datos inequívocos de malignidad de estos tumores son la invasión macroscópica de órganos adyacentes, la presencia de metástasis en los ganglios regionales, hígado o a distancia y la invasión vascular, en la práctica la mayor dificultad reside en predecir el curso de aquellos localizados en el páncreas que, no poseyendo los criterios histológicos generalmente considerados en un tumor maligno, pueden comportarse como tales.
MORFOLOGÍA DE LOS TEP
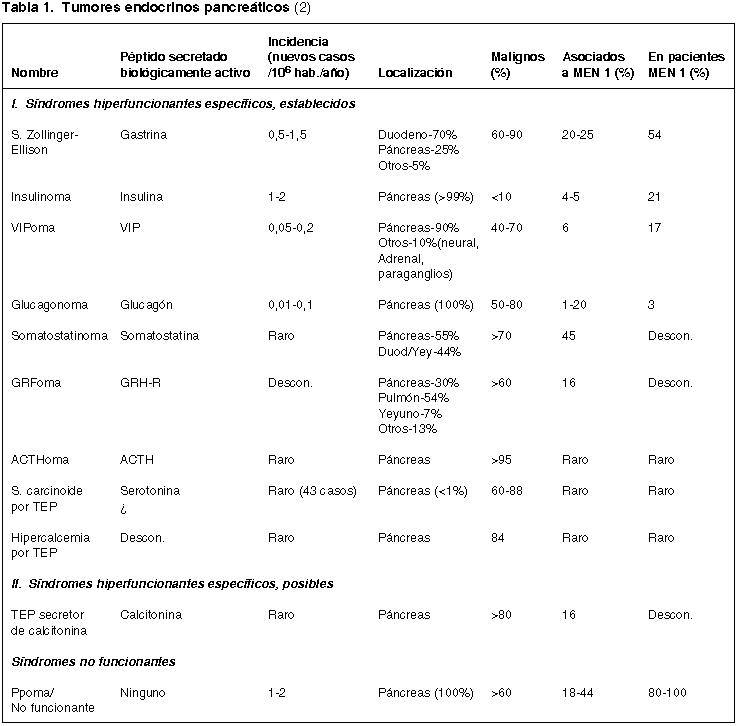
Macroscópicamente son tumores bien delimitados, no rara vez múltiples, sobre todo en casos de MEN 1, con tendencia a localizarse en el cuerpo y la cola del páncreas y que ocasionalmente son quísticos al igual que algunos tumores del páncreas exocrino (1,3) (fig. 1).
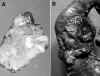
Fig. 1. a) TEP múltiple constituido
por dos nódulos sólidos netamente delimitados. b) TEP con quistificación
central por hemorragia intratumoral.
La mayoría de los TEP tienen una histología con alta diferenciación celular, mostrando diferentes patrones de crecimiento, bien conocidos, que les confiere un aspecto «organoide». No hay necrosis y sus células tienen núcleos isomórficos y sin atipia, con muy escasas mitosis y un citoplasma anfofílico, oxífilo o claro (fig. 2a). A veces tienen estroma amiloide (en PET productores de insulina), melanina, cuerpos de psamoma (en tumores productores de somastotatina) o rara vez osteoide. Pueden estar localizados en el páncreas o presentar metástasis o invasión locoregional en vasos hemáticos, nervios y órganos adyacentes.
Un segundo grupo de TEP, menos frecuente, difiere del anterior en la presencia de cierto grado de atipia y pleomorfismo nuclear, con focos de necrosis y mitosis más frecuentes. El crecimiento organoide es menos aparente y suelen presentar invasión local (fig. 2b).
El tercer y menos frecuente tipo de TEP, corresponde a neoplasias pobremente diferenciadas muy similares en su histología a los carcinomas pulmonares de células pequeñas, con solo una vaga apariencia organoide, muy alta relación núcleo/citoplasmática, «moldeamiento» nuclear, frecuentes células apoptoicas, necrosis geográfica y alto índice mitósico (fig. 2c).
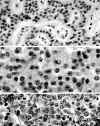
Fig. 2. a) Patrón organoide, de
células isomórficas y típicas (TEP grado 1). b) Atipismo celular discreto con
crecimiento organoide poco aparente (TEP grado 3, carcinoma de bajo grado). c)
Neoplasia de células pequeñas pleomórfica y atípica (TEP grado 4, carcinoma
de células pequeñas).
HISTOQUÍMICA DE LOS TEP
La realización de tinciones argénticas para reconocer la diferenciación neuroendocrina es de escasa utilidad, poco específica, con falsos positivos debidos a la presencia de gránulos citoplasmáticos exocrinos y mucina. El mucicarmín o PAS-diastasa son útiles para separar los TEP de tumores exocrinos o de neoplasias mixtas exocrinas-endocrinas (5,6).
ULTRAESTRUCTURA DE LOS TEP
Ya que los TEP tienen una naturaleza auténticamente neuroendocrina su ultraestructura es característica, con gránulos densos neurosecretorios en su citoplasma. Estos no tienen características particulares para los diferentes neuropéptidos, excepto los correspondientes a insulina, que pueden ser característicos por contener un centro «cristalino» rodeado de un halo submembranoso y los de glucagón con estructura de doble densidad, sin apariencia cristalina (6) (fig. 3).

Fig. 3. Ultraestructura de un TEP
secretor de insulina: Numerosos gránulos electrodensos con centro
"cristalino" rodeado de un ancho halo claro.
El estudio ultraestructural además de demostrar la secreción de insulina y glucagón puede detectar, no infrecuentemente, neoplasias endocrinas con diferenciación pancreática divergente, ya acinar (con gránulos de tipo zimógeno) ya ductal (con diferenciación luminal ductal con microvillis), tumores pancreáticos mixtos exocrino-endocrinos. También puede detectar en adenocarcinomas pancreáticos exocrinos —ductales o acinares— diferenciación neuroendocrina oculta (5,7).
INMUNOHISTOQUÍMICA DE LOS TEP
Como tumores de naturaleza epitelial, los TEP presentan inmunorreactividad para diferentes queratinas, especialmente 8 y 18, coexpresando casi siempre neurofilamentos y a menudo vimentina (8). Tanto la cromogranina A como la sinaptofisina definen su carácter neuroendocrino, con positividad para uno de ellos o para ambos en prácticamente todos los casos (1) (fig. 4). Un hecho descrito de interés, aunque de no mucha importancia práctica, es que los tumores productores de insulina y de somatostatina pueden no ser reactivos para cromogranina A, aunque sí para cromogranina B o C (5).
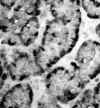
Fig. 4. Positividad intensa para
cromagranina A en el polo basal celular de un TEP bien diferenciado.
¿Es necesario hacer IHQ para demostrar péptidos hormonales específicos, como insulina, glucagón, VIP, etc.? En principio no para aquellos tumores que cursan con cuadros clínicos de endocrinopatía por hipersecreción hormonal, ya que la determinación sérica de los diferentes péptidos es sencilla, constituyendo un procedimiento clínico habitual y la IHQ no es necesaria para el diagnóstico. Sólo en los casos de TEP fisiológicamente «silentes», el marcaje de neuropéptidos específicos puede ser de ayuda; así por ejemplo, la secreción de PP o de somatostatina puede no producir endocrinopatía clínica y su demostración IHQ, de importancia pronóstica, señalará un posible marcador bioquímico para el seguimiento monitorizado del crecimiento tumoral. La existencia de diferencias significativas en la supervivencia media, entre los tumores verdaderamente no funcionantes y los uni y plurihormonales, es discutible cuando los tumores productores de insulina se excluyen del análisis (9).
PATOLOGÍA MOLECULAR DE LOS TEP
Las bases moleculares del desarrollo de los TEP no se conocen con precisión, aunque se sabe que están relacionadas con la vía tumoral supresora. Se conocen mejor en casos relacionados con MEN 1. Estos pacientes frecuentemente presentan una mutación en la línea germinal, en un locus (porción centromérica del brazo largo) del cromosoma 11, gen mu/MEN 1, cuyo producto tiene una función supresora; esta alteración es más rara en casos de TEP esporádico (10,11). También hay datos que sugieren diferente patogénesis molecular según sea un tumor funcionante o no. Las mutaciones del K-ras y CDKN2, frecuentes en los carcinomas del páncreas exocrino, son bastante raras en los TEP.
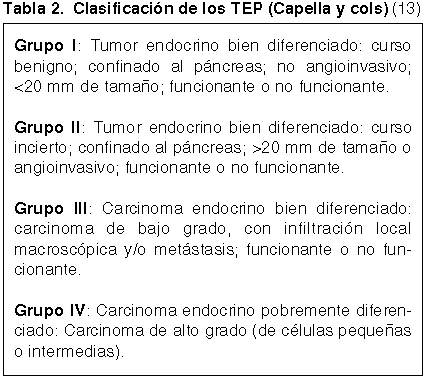
CLASIFICACIÓN MORFOLÓGICA DE LOS TEP
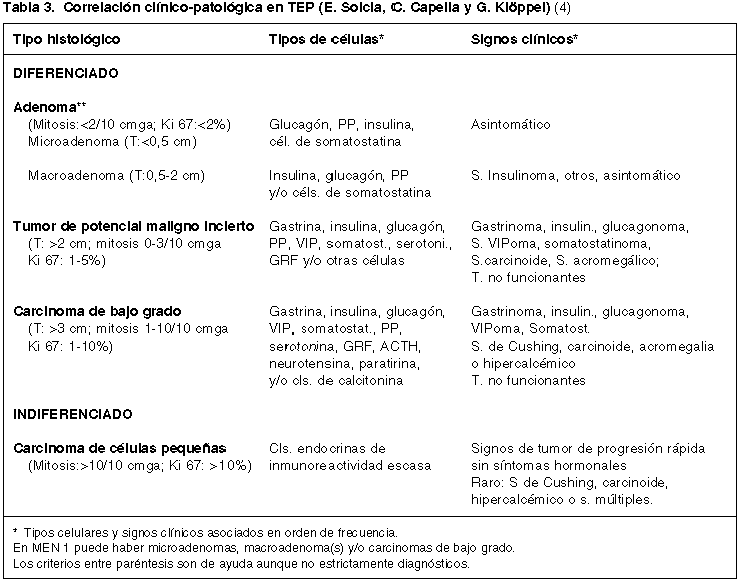
Los TEPs presentan una gradación histológica que es la base fundamental de su clasificación morfológica, que debe completarse con las características clínicas para una más exacta evaluación pronóstica, todavía no conseguida (4,12). La desventaja clínica de las evaluaciones histológicas es su realización post-cirugía con lo que queda limitada su importancia para plantear estrategias terapéuticas. La más aceptada, es la de Capella y cols. (tabla 2), bastante similar a otras también utilizadas, especialmente la de Wick y cols (5,13,14). En ellas se distinguen dos grupos extremos de neoplasias: bien diferenciadas e indiferenciadas. Estas últimas, TEP grado 4, con clara apariencia de un carcinoma de células pequeñas o intermedias y muy similar a su contrapartida pulmonar, con necrosis geográficas y alto índice mitósico (> de 10 mitosis por 10 campos microscópicos de gran aumento, c.m.g.a.). El grupo de neoplasias bien diferenciadas es más heterogéneo, pudiendo delimitarse tumores con aspecto de carcinoma de bajo grado, TEP grado 3, que tienen áreas poco organoides, con pleomorfismo nuclear, áreas de necrosis y mitosis frecuentes, pero en número menor a 10 por 10 c.m.g.a. Son tumores, como los indiferenciados, cuya naturaleza maligna es obvia, pero ambos son poco frecuentes. La gran mayoría de los TEP son neoplasias bien diferenciadas que se han subdividido en dos subgrupos: grado 1 de bajo riesgo o tipo adenoma, TEP grado 1 y grado 1 de riesgo significativo o tipo «borderline» o de significación incierta, TEP grado 2. En estos dos grupos de tumores confinados al páncreas, con apariencia organoide, sin necrosis, se valora la actividad mitósica (0 a 1 mitosis/10 por 10 c.m.g.a. en los de bajo riesgo y 2 a 3 en los de riesgo significativo), pero además datos macroscópicos (tamaño), actividad proliferativa y tipo de secreción-clínica (tabla 3).
DATOS PRONÓSTICOS EN LOS TEP
Es bien conocido que los TEP pueden tener un curso no predecible y que su comportamiento biológico no siempre se correlaciona con sus rasgos morfológicos, especialmente en los tumores bien diferenciados (15) (tabla 4).
a) Datos morfológicos
La agresividad biológica de los TEP está relacionada con el tamaño tumoral (tumores mayores de 2 cm y de 4 cm o más), invasión vascular o perineural e infiltración de la cápsula pancreática, además de con el número de mitosis (>2/10 c.m.g.a.) y obvia atipia nuclear (5,13,16).
b) Secreción tumoral
Las hormonas producidas por los TEP tienen interés pronóstico (4,5,13): De los tumores de bajo grado, los secretores de insulina tienen un riesgo bajo de curso maligno (10%), que llega a ser del 60% para el resto de los TEP. Los tumores que producen péptidos propios del páncreas (insulina<polipéptido pancreático<somatostatina<glucagón) son más frecuentemente benignos que los que producen péptidos entéricos (polipéptido intestinal vasoactivo<gastrina). La producción de neuropéptidos sistémicos (ACTH, calcitonina) se asocia a un curso más agresivo (4). Los tumores no funcionantes son malignos en la mayoría de los casos, aún incluso si son de grado 1 (65%) (9,4,16). No hay una diferencia pronóstica importante entre tumores con secreción uni o plurihormonal.
c) Receptores hormonales
Una característica de las células de los islotes, de significación fisiológica desconocida, posiblemente única entre las células endocrinas del organismo, es que expresan proteínas receptoras de progesterona, reteniendo esta función algunas neoplasias endocrinas pancreáticas (aunque también algunas lesiones exocrinas, como las neoplasias quísticas mucinosas). La ausencia de receptores de progesterona puede ser un signo desfavorable en los TEPs (16,17), aunque está por ver si constituye un dato de riesgo significativo e independiente.
d) Proliferación celular
Existen series publicadas indicando que la actividad proliferativa tumoral detectada con el anticuerpo MIB-1 es un dato de ayuda en la valoración pronóstica de los TEPs de bajo grado, si bien la dificultad de su empleo como marcador de valor pronóstico reside en los problemas metodológicos inherentes a esta técnica y en los diferentes valores de corte que se han utilizado (4,18,19).
e) Otros datos
Hay datos preliminares, aún pendientes de validación, sobre la significación en los TEPs de bajo grado de diferentes proteínas (receptores de laminina, CD44), aneuploídia y diversas alteraciones moleculares (sobrexpresión de K-ras y Ha-ras, LOH para diferentes locus).
En resumen, podemos considerar justificado y conveniente incluir en nuestro informe de un tumor endocrino del páncreas, especialmente si es de bajo grado, los datos morfológicos, inmunohistoquímicos y de proliferación celular (MIB 1) para identificar, evaluados junto a los datos clínicos, la existencia de riesgo significativo de malignidad.
BIBLIOGRAFÍA
Apel RL, Asa SL. Endocrine tumors of the pancreas. Pathol Annu 1995; 30: 305-49.
Jensen RT. Pancreatic endocrine tumors: Recent advances. Ann Oncol 1999; 10 (Suppl 4): S170-6.
Bieligk S, Jaffe BM. Islet cell tumors of the pancreas. Surg Clin North Am 1995; 75: 1025-40.
Solcia E, Capella C, Kloppel G. Tumors of the Pancreas. Atlas of Tumor Pathology, Series 3, Fascicle 20. Washington, DC: Armed Forces Institute of Pathology; 1997. p. 1-105.
Wick MR, Graeme-Cook FM. Pancreatic Neuroendocrine Neoplasms. A current summary of diagnostic, prognostic, and differential diagnostic information. Am J Clin Pathol 2001; 115 (Suppl 1): S28-45.
Larsson LI. Endocrine pancreatic tumors. Hum Pathol 1978; 9: 401-16.
Deshpande V, Selig MK, Nielsen GP et al. Ductulo-insular pancreatic endocrine neoplasms. Clinicopathologic analysis of a unique subtype of pancreatic endocrine neoplasms. Am J Surg Pathol 2003; 27: 461-8.
Kimura N, Nakazato Y, Nagura H, et al. Expression of intermediate filaments in neuroendocrine tumors. Arch Pathol Lab Med 1990; 114: 506-10.
Lo CY, van Heerden JA, Thompson GB, et al. Islet cell carcinoma of the pancreas. World J Surg 1996; 20: 878-84.
Debelenko LV, Zhuang Z, Emmert-Buck MR, et al. Allelic deletions on chromosome 11q13 in multiple endocrine neoplasia typ1-1 associated and sporadic gastrinomas and pancreatic endocrine tumors. Cancer Res 1997; 57: 2238-43.
Bergman L, Boothroyd C, Palmer J, Grimmond S, et al. Identification of somatic mutations of the MEN 1 gene in sporadic endocrine tumours. Br J Cancer 2000; 83: 1003-8.
Schindl M, Kaczirek K, Kaserer K, Niederle B Is the new classification of neuroendocrine pancreatic tumors of clinical help? World J Surg 2000; 24: 1312-8.
Capella C, Heitz PU, Hofler H, et al. Revised classification of neuroendocrine tumours of the lung, pancreas, and gut. Virchows Arch 1995; 425: 547-60.
Wick MR. Neuroendocrine neoplasia: current concepts. Am J Clin Pathol 2000; 113: 331-5.
Lam KY, Lo CY. Pancreatic endocrine tumour: a 22-year clinico-pathological experience with morphological, immunohistochemical observation and a review of the literature. Eur J Surg Oncol 1997; 23: 36-42.
La Rosa S, Sessa F, Capella C, et al. Prognostic criteria in nonfunctioning pancreatic endocrine tumours. Virchows Arch 1996; 429: 323-33.
Viale G, Doglioni C, Gambacorta M, et al. Progesterone receptor immunoreactivity in pancreatic endocrine tumors: an immunocytochemical study of 156 neuroendocrine tumors of the pancreas, gastrointestinal and respiratory tracts, and skin. Cancer 1992; 70: 2268-77.
Perret GA, Mosnier JF, Buono JP, et al. Relationship between MIB-1 proliferation index and outcome in pancreatic neuroendocrine tumors. Am J Clin Pathol 1998; 109: 286-93.
Lloid RV. Utility of Ki-67 as a prognostic marker in pancreatic endocrine neoplasms. Am J Clin Pathol 1998; 109: 245-7.
![]()