
Vol. 37, n.º 1, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 1, 2004 |
Pilar Molina Aguilar1, Concepción Dasí Martínez2, Marina S. Gisbert Grifo3
1 Servicio de Patología. Instituto de
Medicina Legal de Valencia.
2 Sección de Histopatología. Instituto de Toxicología de
Barcelona.
3 U.D. de Medicina Legal. Departament de M.P. i S.P., Brom., Tox. i
Medicina Legal. Facultat de Medicina i Odontología. Universitat de Valencia
E.G.
molina_pil@gva.es
RESUMEN
En Patología Forense, frecuentemente nos encontramos con autopsias de muertes súbitas en las que se observan lesiones inespecíficas (edema pulmonar y/o cerebral) como únicos hallazgos macroscópicos y en ocasiones también microscópicos. La información clínica suele ser escasa, la muerte normalmente ocurre sin testigos y el lugar del levantamiento aporta pocos datos orientativos. Nos encontramos, en principio, ante la llamada incorrectamente «autopsia blanca». En estos casos, es imprescindible descartar la participación de tóxicos o trastornos electrolíticos, así como estar familiarizados con todas aquellas patologías que puedan cursar con una muerte súbita. La casuística en este tipo de muertes es reducida debido a su escasa frecuencia y a la dificultad de inscribir correctamente la causa de muerte en los registros de mortalidad. Este artículo trata de revisar aquellas patologías extracardíacas que, con mayor frecuencia, nos podemos encontrar en este tipo de autopsias: muerte súbita e inesperada del epiléptico, trauma cerebral mínimo, asma bronquial, hipertensión pulmonar, microembolismo, diabetes mellitus tipo I y cetoacidosis alcohólica. En muchos de estos casos, los estudios químico-toxicológicos e histológicos postmortem pueden establecer la causa de muerte, si los datos clínicos y los obtenidos en el momento del levantamiento son compatibles con la misma.
Palabras Clave: Autopsia blanca, patología forense, muerte súbita.
SUMMARY
Sudden death autopsy is a frequent event in forensic pathology. In a percentage of these cases, unspecific pathology (pulmonary and/or cerebral edema) is the only feature observed in postmortem examination. Deaths are usually witnessed. Clinical data, scene information and circumstances of death are insufficient. The aim of this paper is to review the most frequent extracardiac diseases that could result in a negative autopsy: SUDEP (sudden unexplained death in epilepsy), bronchial asthma, minimal traumatic brain injury, pulmonary hypertension, microembolism (fat, amniotic fluid and air embolism), type I diabetes mellitus (dead in bed syndrome) and alcoholic ketoacidosis. Laboratory tests and careful histological examination may establish the cause of death in cases where clinical and scene data are compatible.
Key words: Negative autopsy, forensic pathology, sudden death
En Patología Forense, frecuentemente nos encontramos con autopsias de muertes súbitas o cuyas circunstancias se desconocen, en las que sólo se observan lesiones inespecíficas (congestión visceral generalizada, edema pulmonar y/o cerebral) como únicos hallazgos macroscópicos y en ocasiones también microscópicos. En general, la información clínica suele ser escasa, la muerte normalmente ocurre sin testigos y el lugar del levantamiento aporta pocos datos orientativos. Nos encontramos, en principio, ante la llamada incorrectamente «autopsia blanca». En estos casos, es imprescindible descartar la participación de tóxicos o trastornos electrolíticos mediante el estudio toxicológico y bioquímico de muestras de sangre, orina y humor vítreo, así como estar familiarizados con todas aquellas patologías que puedan cursar con una muerte súbita, incluso como síntoma inicial.
La casuística en este tipo de muertes es reducida debido a su escasa frecuencia y a la dificultad de inscribir correctamente la causa de muerte en los certificados médicos de defunción y en el boletín estadístico de defunción, y por tanto en los registros de mortalidad (1).
Este artículo trata de revisar aquellas patologías que, con mayor frecuencia, nos podemos encontrar en este tipo de autopsias, exceptuando las de origen cardíaco, detalladas en otro artículo de esta misma monografía. En muchos de estos casos, los estudios químico-toxicológicos e histológicos postmortem pueden establecer la causa de muerte, si los datos clínicos y los obtenidos en el momento del levantamiento son compatibles con la misma.
MUERTE SÚBITA E INESPERADA EN EPILEPSIA
La muerte súbita e inesperada del epiléptico (Sudden Unexpected Death in Epilepsy, SUDEP) fue definida en 1997 por el grupo de expertos como «muerte súbita, inesperada, con o sin testigos, no traumática y no por ahogamiento, en un paciente epiléptico, con o sin evidencia de que haya sufrido una crisis epiléptica al morir, en el que se haya descartado un estado convulsivo como causa de muerte y en el que la autopsia no proporcione evidencia alguna de una causa anatómica o tóxica de la muerte» (2,3).
De la anterior definición y de posteriores trabajos (4-6), derivan una serie de criterios necesarios para el diagnóstico definitivo de SUDEP:
1. La víctima debe tener un diagnóstico previo de epilepsia
2. La muerte debe ocurrir inesperadamente en un paciente con buen estado de salud y de forma súbita (si hay testigos)
3. Puede o no haber evidencia de una crisis epiléptica al morir (lengua, labios o mucosa yugal mordidos) pero nunca de un status epiléptico.
4. En la autopsia debe descartarse cualquier causa de muerte accidental (traumatismo, sumersión, intoxicación y aspiración) o natural (pe. patología cardiorrespiratoria importante).
Si se cumplen todos los criterios clínicos pero no se realiza autopsia, debe incluirse dentro de la categoría de probable SUDEP y si existe una alta sospecha clínica pero faltan evidencias y el estudio necrópsico, en posible SUDEP.
Su incidencia se estima en un 8-17% de las muertes en epilépticos y su frecuencia aumenta en epilepsias refractarias al tratamiento. Es más frecuente en hombres (7:4) en la tercera y cuarta décadas de la vida, con un pico máximo a los 28-35 años. Las circunstancias de la muerte suelen ser desconocidas, puesto que en la mayoría de los casos ocurre sin testigos o durante la noche. Dentro de los factores de riesgo se citan (7-11): crisis tónico-clónicas generalizadas, déficit neurológico, alcoholismo, politerapia, escaso control médico, frecuentes ajustes de dosis de antiepilépticos, tratamiento concomitante con antipsicóticos, epilepsias de comienzo precoz y de más de 10 años de evolución y crisis sin testigos.
Todos los estudios epidemiológicos apoyan la crisis epiléptica como factor causal de la muerte, que produciría un fracaso cardiorrespiratorio ictal o postictal inducido por los cambios fisiológicos demostrados durante las crisis (12). El edema pulmonar neurógeno (13), la apnea central (14,15) y la arritmia cardiaca (16-18), inducidos por una descarga a-adrenérgica de origen central, constituyen los tres mecanismos fisiopatogénicos más estrechamente relacionados con la SUDEP (fig. 1).
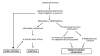
Fig. 1. Mecanismos fisiopatogénicos
postulados en la SUDEP.
Los hallazgos autópsicos en este tipo de muerte son inespecíficos e incluyen:
1. Cerebrales: en la mayoría de los casos existe patología crónica (10,19-21): esclerosis mesial, malformaciones vasculares, enfermedades degenerativas, infartos o contusiones antiguas y patología aguda, como se ha demostrado en un trabajo reciente (22), mediante inmunoexpresión de HSP-70 y c-Jun en el hipocampo de los casos de SUDEP. La presencia de lesión neuronal aguda y su distribución sugieren una muerte no instantánea y posterior a una crisis epiléptica.
2. Pulmonares: se observa edema en grado moderado o severo en un 60-80% de los casos. El peso de los pulmones suele estar aumentado en un 110-190% del normal e histológicamente se demuestra un edema alveolar denso y hemorrágico (23). En algunos casos, el edema puede ser lo suficientemente severo como para establecerse como causa de muerte (aumento del peso en un 200-375% del normal) (24).
3. Cardiacos: se han descrito signos de isquemia crónica subendocárdica en ausencia de arterioesclerosis coronaria, que según postulan los autores de dicho trabajo, serían causados por vasoespasmos coronarios de origen central asociados a las crisis y podrían justificar un origen arrítmico cardiaco de la SUDEP (25). Sin embargo, en otro estudio similar no se encuentran diferencias entre los casos de SUDEP y los casos control, incluido el estudio del sistema de conducción cardiaco (26). Puesto que en ambos trabajos, el número de casos es reducido (7 y 10 respectivamente), por el momento no parecen existir datos concluyentes respecto a la patología cardiaca asociada a SUDEP (27,28).
4. Hepáticos: en la mayoría de SUDEP se observan signos de fallo cardiaco derecho (aumento del peso de la víscera y congestión crónica) (9,20).
5. Toxicológicos: en un alto porcentaje de casos se detectan niveles subterapéuticos de los fármacos antiepilépticos (19,29,30).
Es importante realizar un estudio autópsico exhaustivo en estos casos, para establecer el diagnóstico de SUDEP e inscribirlo como tal en los registros de mortalidad. Esta información es necesaria para los estudios multicéntricos destinados a identificar su incidencia (31), posibles factores de riesgo o mecanismos fisiopatológicos implicados. En este momento, los datos epidemiológicos indican que disminuir médicamente la frecuencia de las crisis y cambiar la postura, estimulando la respiración durante la crisis pueden evitar la SUDEP. Estas recomendaciones se basan en estudios comparativos con colectivos de epilépticos con una alta supervisión o con crisis en presencia de testigos (19,31,32). Los resultados de un trabajo español, aunque de muestra reducida, son muy similares (33).
Por último, si en cualquier autopsia es imprescindible la información clínica, en estos casos, lo es aún más, puesto que existe un síndrome arrítmico que puede debutar con muerte súbita y que clínicamente simula un cuadro epiléptico: el síndrome de QT largo. Existe un número no desdeñable de casos descritos en la literatura médica que fueron diagnosticados de epilepsia cuando estudios cardiacos posteriores o retrospectivos demostraron que realmente se trataban de síndromes de QT largo (3). La llamada autopsia molecular permitirá en un futuro descartar este síndrome de forma rutinaria (34). Por el momento, deberíamos completar el protocolo de autopsia de SUDEP con la revisión de posibles ECG realizados en vida y congelar tejido miocárdico, hepático o esplénico para posibilitar futuros estudios moleculares (35).
LESIÓN TRAUMÁTICA CEREBRAL MÍNIMA
Black y Graham (21) describen en una reciente revisión de causas de muerte súbita por patología intracraneal la existencia de casos de muertes súbitas cuyas circunstancias sugieren un origen traumático y en los que sólo encuentran lesiones traumáticas mínimas en tórax y abdomen, o sólo a nivel craneal, que no justifican la causa de muerte.
Aunque uno esperaría encontrar la causa de la muerte en el estudio macroscópico y/o microscópico cerebral, tan sólo se encuentran lesiones externas demostrativas de un traumatismo, como focos de hemorragia subdural con mínima contusión cerebral superficial, sin existir afectación intraparenquimatosa contusiva o hemorrágica.
En algunos casos, la rapidez del proceso letal impide identificar lesiones a nivel histológico, pudiendo ser consecuencia de una severa disfunción neuroquímica o autonómica de origen central, similar a la demostrada en las roturas de aneurismas saculares intracraneales. En estos casos podemos encontrar hemorragias subendocárdicas en sábana en ventrículo izquierdo de localización subvalvular aórtica predominante, indicando un posible fallo cardíaco arrítmico próximo a la muerte, o una ingesta elevada de alcohol que agrava el potencial lesivo del traumatismo (36).
En otros casos, el examen macroscópico cerebral post-fijación puede revelar un infiltrado hemorrágico petequial difuso que tiende a localizarse en la sustancia blanca de las porciones anteriores de los lóbulos frontal y temporal y en las estructuras periventriculares de la base cerebral. Esta lesión (daño vascular difuso) es considerada como una forma de daño cerebral difuso post-traumático (37) y parece observarse sólo cuando la muerte es muy inmediata al trauma craneal.
El daño axonal difuso no suele cursar con muertes inmediatas al traumatismo, aunque sería posible identificarlo en algunos cadáveres en los que se desconocen las circunstancias de la muerte. En este sentido, la identificación inmunohistoquímica de la proteína precursora del ß-amiloide (ß-APP) permite diagnosticar la lesión axonal aguda más precozmente (2-3h) que las técnicas de tinción convencionales (12-18h). Según las situaciones, el diagnóstico diferencial deberá incluir la hipoxia cerebral global, la encefalitis vírica y el embolismo graso (38).
ASMA BRONQUIAL
El asma, junto con las hemorragias intracraneales y la epilepsia, constituyen las causas mas frecuentes de muerte súbita extracardíaca en la población menor de 36 años (1). Normalmente la muerte sobreviene en un status asmático con o sin tratamiento hospitalario, pero también, aunque más raramente, puede ocurrir de forma súbita, tras una crisis asmática (con un intervalo de 3-12h), durante o inmediatamente después de la crisis o por la noche durmiendo, en cuyo caso, es encontrado muerto en la cama al día siguiente (39).
La patología asmática es fácilmente identificable en autopsias de muertes acontecidas durante un status asmático o inmediatamente posterior al mismo. Pero estos hallazgos macroscópicos típicos, hiperinsuflación pulmonar y tapones de moco intrabronquiales, pueden pasar mas desapercibidos en autopsias donde se desconocen los antecedentes personales e inmediatos a la muerte o ser de menor intensidad en los casos de muerte súbita asociada o no a una crisis asmática. En estos casos, la histología pulmonar es claramente demostrativa de un proceso asmático, aunque no distinguible como causa inmediata de muerte.
Existen estudios recientes dirigidos a diferenciar histológicamente un ataque agudo del asma crónico:
Infiltrado inflamatorio submucoso: predominio de neutrófilos sobre eosinófilos (40,41), menor número de mastocitos íntegros pero mayor número de mastocitos degranulados (42) y mayor número de basófilos (cuantificados mediante técnicas inmunohistoquímicas: 2D7) (43) en muertes súbitas (menos de 1 h desde el comienzo de los síntomas).
Secreción mucosa: mediante la medición de áreas, se observa mayor obstrucción bronquial por moco y mayor área de glándulas mucosas en los casos de muerte súbita en un ataque agudo (42).
También se ha estudiado la expresión de lactoferrina (41), mediador de la inflamación en vías aéreas, y la cuantificación de fibras elásticas en submucosa (44). Todos estos estudios concluyen en el papel esencial de la correlación clínico-patológica para interpretar los resultados.
La causa de la muerte súbita en asmáticos permanece como interrogante, aunque se han implicado mecanismos de tipo asfíctico, arrítmico y metabólico (hipopotasemia) relacionados con el tratamiento con ß-agonistas. En este sentido, existen trabajos contradictorios:
Los trabajos que implican la cardiotoxicidad e hipopotasemia producida por los fármacos ß-agonistas en la causa de muerte, se apoyan en su conocida acción de prolongación del intervalo QT (45) y los niveles postmortem elevados de estos fármacos, posiblemente producidos por su autoadministración creciente durante las crisis (46).
Por el contrario, los autores de los trabajos que abogan por un mecanismo asfíctico, donde la hipoxia y acidosis producirían una arritmia maligna, se basan en estudios retrospectivos clínicos (47) o autópsicos (48). En estas series, se demuestra como factor determinante la severidad del asma (que explicaría las dosis aumentadas de ß-agonistas), la subestimación de gravedad de la crisis y las infecciones víricas concomitantes.
Una explicación adicional que integra ambas, sería la propuesta por Burggraaf y cols (49): los fármacos ß-agonistas administrados en estados hipóxicos producirían una depresión cardiorrespiratoria por reflejo vagal mediado por la disminución del retorno venoso producido por vasodilatación periférica y shunt pulmonar. Explican con ello, el predominio de muertes súbitas en asmáticos no hospitalizados, automedicados y sin oxigenoterapia concomitante.
Ante una autopsia de muerte súbita en un asmático, y por tanto no asociada a un status asmático, debe considerarse un mecanismo mixto de tipo asfíctico-arrítmico como causa de muerte, en ausencia de otros hallazgos patológicos.
HIPERTENSIÓN PULMONAR
La hipertensión pulmonar primaria afecta típicamente a mujeres jóvenes (20-40 años). En su forma familiar se ha identificado el locus cromosómico 2q31-q32. Puede debutar con muerte súbita como primer síntoma (50-54) en aproximadamente un 5% (55) de los casos, citándose como factores precipitantes el estrés y el embarazo (3er trimestre y puerperio precoz) (56).
El mecanismo de muerte parece ser multifactorial incluyendo el efecto arritmogénico de la hipertrofia ventricular derecha y la bradicardia inducida por la hipoxia (55). Se ha descrito también la compresión del tronco principal de la arteria coronaria izquierda por la dilatación de la arteria pulmonar como causa de isquemia miocárdica (57).
Los datos de autopsia pueden revelar una hipertrofia ventricular derecha, dilatación del cono pulmonar, placas de ateroma en arterias pulmonares y signos sistémicos de fallo cardiaco derecho.
Si nos encontramos con una hipertensión pulmonar primaria como causa de muerte súbita, debemos tener presentes las posibles implicaciones familiares y notificarlo pertinentemente. La hipertensión pulmonar secundaria también puede cursar con muerte súbita, pero en estos casos la autopsia normalmente revelará la patología causante de la misma.
MICROEMBOLISMO
Embolismo graso
El síndrome de embolismo graso no ofrece problemas a la hora de diagnosticarlo tanto clínica como histológicamente (H&E). El problema puede surgir si:
El paciente muere en la fase hiperaguda del mismo, por un fallo cardíaco derecho agudo de tipo obstructivo, sin dar tiempo a que se observen los signos típicos sistémicos de la lesión vascular en la autopsia (rash petequial cutáneo y severo infiltrado petequial cerebral).
El embolismo se desarrolla en el seno de patologías, pe. drepanocitosis (58,59), diagnósticos clínicos, pe. shock traumático, politraumatismos de partes blandas sin fracturas (60,61) o intervenciones quirúrgicas no asociadas frecuentemente con el mismo, pe. artrodesis de columna vertebral (comunicación personal del autor, XX Congreso Nacional de la Sociedad Española de Anatomía Patológica).
En caso de no observarse embolismo sistémico, según la magnitud del mismo a nivel pulmonar y la patología previa del paciente, se puede establecer éste como causa de muerte inmediata o como concausa.
Bajo nuestra experiencia, creemos necesaria la realización de técnicas histoquímicas especificas (oil red u osmio) para establecer, mediante un score fiable, la cantidad de grasa embolizada, ya que la valoración morfológica puede ser errónea (60,62). Siempre es necesaria la correlación clínicopatológica y descartar el origen yatrogénico por maniobras de reanimación cardiopulmonar.
Embolismo de líquido amniótico
El embolismo de liquido amniótico tiene una incidencia muy baja (1/8.000-80.000 embarazos) pero una mortalidad elevada (61-86%) (63). Aunque es más frecuente en el periodo periparto, se han descrito casos en el segundo trimestre (64).
En la autopsia, es primordial el estudio pormenorizado del útero, incluyendo cérvix (65), placenta y membranas. El diagnóstico definitivo se establece al demostrar la presencia de componentes del líquido amniótico en arterias y capilares pulmonares. Aunque generalmente es fácil detectar estos componentes principales (escamas epiteliales, grasa del vernix caseoso, mucinas del meconio), puede ser útil la utilización de luz polarizada (lanugo fetal) (56) y la realización de técnicas histoquímicas (detección de grasa) e inmunohistoquímicas, como queratinas de alto peso molecular (56), TKH-2 (mucina derivada de meconio y líquido amniótico) (66), beta-hCG y hPL (67) o antígenos fetales (68).
La presencia de embolismo pulmonar de líquido amniótico se ha demostrado en pacientes durante el período postparto sin clínica evidente (56). Este hecho, junto con la demostración experimental de la inocuidad de la administración intravenosa de líquido amniótico en animales y humanos (69), indican la falta de criterios diagnósticos específicos de este síndrome y la necesidad de una presentación clínica compatible (70).
Las últimas investigaciones sugieren como mecanismo fisiopatológico, una respuesta inmune materna frente a los antígenos fetales, de tipo anafiláctico (71), anafilactoide (69) o mediada por el complemento (72). Este dato aporta posibles criterios diagnósticos adicionales para poder establecer el embolismo de líquido amniótico como causa de muerte:
Fineschi y cols (1998) (73), mediante técnicas inmunohistoquímicas (triptasa) y morfométricas, demuestran un aumento de mastocitos pulmonares en los seis casos estudiados de muerte por embolismo de líquido amniótico.
Nishio y cols (2002) (74) identifica niveles séricos elevados de triptasa postmortem en un caso con diagnóstico histológico de embolismo de líquido amniótico.
Rainio y col (2003) (75) presentan un caso de muerte súbita por embolismo de líquido amniótico tras un accidente de tráfico, y demuestran la activación de mastocitos en el pulmón mediante inmunotinción de mastocitos degranulados con triptasa.
Embolismo aéreo
La autopsia en caso de un embolismo aéreo es típicamente negativa. Aunque no se sospeche clínicamente, debe descartarse en autopsias de muertes súbitas intraoperatorias o en pacientes portadores de catéteres venosos. La realización de técnicas radiológicas previas (Rx, TAC, RNM) es imprescindible en este tipo de autopsias (62,76,77). La demostración de aire en cavidades cardíacas derechas debe hacerse mediante disección bajo agua y en arterias meníngeas mediante un examen cerebral externo in situ y posterior disección cerebral bajo agua (78,79). El volumen de aire en el sistema venoso necesario para causar la muerte se estima en 100-250 ml, sin embargo, cantidades mucho mas pequeñas pueden causar la muerte en el embolismo arterial (79). Mediante técnicas especiales es posible aspirar el gas embolizado y analizar sus componentes (78). Las maniobras de reanimación cardiopulmonar o un intervalo postmortem elevado interfieren e imposibilitan el diagnóstico de embolismo aéreo.
Aunque la documentación de los hallazgos histológicos es escasa, la mayoría de los autores coinciden en interpretar como un signo de vitalidad la presencia de espacios aéreos rodeados de leucocitos y plaquetas. Este fenómeno es explicado y demostrado experimentalmente por Ritz-Timme y cols (1998) (80) por la interacción de fenómenos fisicoquímicos entre ambos componentes (células y aire) mientras existe circulación sanguínea.
Puesto que el foramen oval persistente tiene una prevalencia de aproximadamente el 30% de la población (81), ante cualquier tipo de embolismo es importante constatar si existe y descartar una embolia paradójica.
DIABETES MELLITUS TIPO I
En 1991, Tattersall y Gill (82) publican, a raíz de un estudio sobre 50 casos de muerte súbita e inexplicada en diabéticos tipo I menores de 50 años, una serie de 22 casos en los que la autopsia no revela ninguna causa de muerte y que reúnen unas características comunes: edad comprendida entre 12 y 43 años, la muerte tiene lugar durante la noche, siendo encontrados al día siguiente en la cama en una apariencia normal y como único antecedente previo: episodios de hipoglucemia nocturnos en los 6 meses anteriores en más de la mitad de los casos.
Este fenómeno se empezó a conocer como «dead in bed syndrome» (83) publicándose casos similares por otros autores (84-87). Actualmente se calcula una incidencia aproximada del 6% del total de las muertes en diabéticos menores de 40 años (88).
La hipoglucemia nocturna se ha establecido como factor precipitante, basándose en estudios clínicos, puesto que la autopsia no aporta datos específicos. El diagnóstico de hipoglucemia es difícil de confirmar por los posibles fenómenos de glucolisis y glucogenolisis postmortem (87), aunque recientes estudios en humor vítreo y líquido sinovial combinando más parámetros (lactato, fructosamina, hemoglobina glicosilada) están demostrando que se puede predecir con mayor exactitud el nivel de glucosa antemortem (89-91).
Se ha asociado la neuropatía diabética autónoma (92-94) como factor de riesgo, que contribuiría a la aparición de arritmias ventriculares letales en estados hipoglucémicos. Sin embargo, los trabajos de Heller y cols (95-98) apoyan la existencia de una alteración de la repolarización miocárdica independiente de la neuropatía y directamente relacionada con la hipoglucemia: han demostrado que la hipoglucemia inducida por la administración exógena de insulina produce una forma adquirida del síndrome QT largo, posiblemente a través de la acción simpaticoadrenérgica que regula, vía AMPc, los canales de potasio, calcio y cloro. Estos hallazgos permiten una posible vía terapéutica con fármacos ß-bloqueantes, identificando a aquellos pacientes con alto riesgo de alargamiento del intervalo QT en estados hipoglucémicos (97).
No obstante, debemos ser muy cautos en el diagnóstico de éste síndrome, y disponer de datos bioquímicos postmortem fiables, puesto que como demuestran Edge y cols (1999) (87), en una serie de 116 muertes en diabéticos menores de 20 años, de los nueve casos de posible «dead in bed syndrome», se demostró como causa de muerte una cetoacidosis diabética en tres, una miocarditis focal en uno y un coma hipoglucémico en dos.
CETOACIDOSIS ALCOHOLICA
La muerte súbita en el alcoholismo crónico, se describió por primera vez en 1926 por LeCount y Singer (99), asociada a una severa esteatosis hepática (único hallazgo en las autopsias de los 11 casos). Hoy, resulta interesante leer la perspectiva histórica de este artículo de Chejfec (2001) (100), donde se exponen los avances descritos desde entonces.
En los últimos años se han seguido describiendo casos similares (101-104):
Hallazgos macroscópicos inespecíficos en la autopsia (edema pulmonar y cerebral, cardiomegalia con dilatación de cavidades, gastritis erosiva y esteatosis hepática)
Análisis microbiológicos y toxicológicos negativos o únicamente niveles bajos de alcoholemia postmortem.
En estos casos se ha demostrado niveles altos de ß-hidroxibutarato en sangre, orina y humor vítreo, pudiendo establecerse una cetoacidosis alcohólica como causa de muerte si clínicamente es compatible. En la mayoría, la investigación retrospectiva demostró periodos de abstinencia previos al fallecimiento por diferentes causas.
Se han implicado también fenómenos arrítmicos (105), pero parecen estar más relacionados con la abstinencia tras un consumo irregular pero abusivo del mismo (106-108).
CONCLUSIONES
La autopsia sigue siendo un elemento válido e imprescindible en la investigación clínica. Aunque en las muertes súbitas generalmente se produce un fenómeno arrítmico letal no identificable a nivel histopatológico, el estudio autópsico exhaustivo con técnicas radiológicas, químico-toxicológicas y anatomopatológicas, junto con una información clínica completa, constituye un elemento fundamental en la investigación de este tipo de muertes.
En este sentido, es necesaria una estrecha colaboración entre clínicos, patólogos y médicos forenses que favorecería la posible identificación de factores de riesgo, patologías asociadas o la demostración de resultados compatibles con las hipótesis clínicas.
BIBLIOGRAFÍA
Morentín B, Suárez-Mier MP, Audicana C, Aguilera B, Garamendia PM, Elexped X. Incidencia y causas de muerte súbita en menores de 36 años. Med Clin (Barc) 2001; 116: 281-5.
Nashef L. Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia 1997; 38 (Suppl 11): S68.
Garaizar C. Muerte súbita, inesperada e inexplicable, en epilepsia. Rev Neurol 2000; 31: 436-41.
Annegers JF, Coan SP, Hauser WA, Leestma J. Epilepsy, vagal nerve stimulation by the NCP system, all-cause mortality, and sudden, unexpected, unexplained death. Epilepsia 2000; 41: 549-53.
Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry 2000; 68: 211-13.
Sperling MR. Sudden Unexplained Death in Epilepsy. Epilepsy Currents 2001; 1: 21-3.
Nashef L, Fish DR, Sander JW, Shorvon SD. Incidence of sudden unexpected death in an adult outpatient cohort with epilepsy at a tertiary referral centre. J Neurol Neurosurg Psychiatry 1995; 58: 462-4.
Lhatoo SD, Langan Y, Sander JWAS. Sudden unexpected death in epilepsy. Postgrad Med J 1999; 75: 706-9.
Langan Y. Suden unexpected death in epilepsy (SUDEP): risk factors and case control studies. Seizure 2000; 9: 179-83.
Antoniuk SA, Oliva LV, Bruck I, Malucelli M, Yabumoto S, Castellano JL. Sudden unexpected, unexplained death in epilepsy autopsied patients. Arq Neuropsiquiatr 2001; 59: 40-5.
Opeskin K, Berkovic SF. Risk factors for sudden unexpected death in epilepsy: a controlled prospective study based on coroners cases. Seizure 2003; 12: 456-64.
Baumgartner C, Lurger S, Leutmezer F. Autonomic symptoms during epileptic seizures. Epileptic Disorders 2001; 3: 103-16.
Johnston SC, Horn JK, Valente J, Simon RP: The role of hypoventilation in a sheep model of epileptic sudden death. Ann Neurol 1995; 37: 531-7.
Nashef L, Walker F, Allen P, Sander JWAS, Shorvon SD, Fish DR: Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry 1996; 60: 297-300.
So EL, Sam MC, Lagerlund TL: Postictal central apnea as a cause of SUDEP: evidence from near-SUDEP incident. Epilepsia 2000; 41: 1494-7.
Massetani R, Strata G, Galli R: Alteration of cardiac function in patients with temporal lobe epilepsy: different roles of EEG-ECG monitoring and spectral analysis of RR variability. Epilepsia 1997; 38: 363-9.
Nei M, Ho RT, Sperling MR: EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia 2000; 41: 542-8.
Rocamora R, Kurthen M, Lickfett L, von Oertzen J, Elger CE. Cardiac asystole in epilepsy: clinical and neurophysiologic features. Epilepsia 2003; 44: 179-86.
Kloster R, Engelskjon T. Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry 1999; 67: 439-44.
Earnest MP, Thomas GE, Eden RA. The sudden unexplained death syndrome in epilepsy: demographic, clinical and postmortem features. Epilepsia 1992; 33: 310-6.
Black M, Graham DI. Sudden unexplained death in adults caused by intracranial pathology. J Clin Pathol 2002; 55: 44-50.
Thom M, Seetah S, Sisodiya S, Koepp M, Scaravilli F. Sudden and unexpected death in epilepsy (SUDEP): evidence of acute neuronal injury using HSP-70 and c-Jun immunohistochemistry. Neuropathol Appl Neurobiol 2003; 29: 132-43.
Terrence CF, Rao GR, Perper JA: Neurogenic pulmonary edema in unexpected, unexplained death of epileptic patients. Ann Neurol 1981; 9: 458-64.
Schnabel R, May TH, Rambeck B. Post-mortem determination of anticonvulsant concentrations and lung oedema in the sudden, unexpected death of epileptics [letter]. Seizure 1997; 6: 327-28.
Natelson BH, Suarez RV, Terrence CF, Turizo R. Patients With Epilepsy Who Die Suddenly Have Cardiac Disease. Arch Neurol 1998; 55: 857-60.
Opeskin K, Thomas A, Berkovic SF. Does cardiac conduction pathology contribute to sudden unexpected death in epilepsy. Epilepsy Res 2000; 40: 17-24.
Langan Y, Sander JWAS. Patients with epilepsy who die suddenly do not always have cardiac disease. Arch Neurol 1999; 56: 249.
Langan Y, Nashef L. Suden unexpected death in epilepsy (SUDEP). ACNR 2003; 2: 6-8.
Vickery BG. Mortality in a consecutive cohort of 248 adolescents who underwent diagnostic evaluation for epilepsy surgery. Epilepsia 1997; 38: S67-9.
George GR, Davis GG. Comparison of anti-epileptic drug levels in different cases of sudden death. J Forensic Sci 1998; 43: 598-603.
Langan Y, Nashef L, Sander JWAS. Certification of deaths attributable to epilepsy. J Neurol Neurosurg Psychiatry 2002; 73: 751-2.
Nashef L. From mystery to prevention: sudden unexpected death in epilepsy, time to move on. J Neurol Neurosurg Psychiatry 1999; 67: 427.
Morentin B, Alcaraz R. Muerte súbita inesperada en epilepsia en niños y jóvenes. Rev Neurol 2002; 34: 462-5.
Ackerman MJ, Tester DJ, Driscoll DJ. Molecular autopsy of sudden unexplained death in the young. Am J Forensic Med Pathol 2001; 22: 105-11.
Cohle SD, A Barbara. The negative autopsy: Sudden cardiac death or other? Cardiovascular Pathology 2001; 10: 219-22.
Milanovic AV, DiMaio VJ. Death due to concussion and alcohol. Am J Forensic Med Pathol 1999; 20: 6-9.
Graham DI, Gennarelli TA, Mcintosh TK. Trauma. En: Graham DI, Lantos PL, editors. Greenfields neuropathology. 7.ª ed. London: Edward Arnold; 2002. p. 823-82.
Geddes JF, Whitwell HL, Graham DI. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol 2000; 26: 105-16.
Champ CS, Byard RW. Sudden death in asthma in childhood. Forensic Sci Int 1994; 66: 117-27.
Sur S, Crotty TB, Kephart GM, Hyma BA, Colby TV, Reed CE et al. Sudden-onset fatal asthma. A distinct entity with few eosinophils and relatively more neutrophils in the airway submucosa?. Am Rev Respir Dis 1993; 148: 713-9.
Tsokos M, Paulsen F. Expression of pulmonary lactoferrin in sudden-onset and slow-onset asthma with fatal outcome. Virchows Arch 2002; 441: 494-9.
Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax 2002; 57: 677-82.
Kepley CL, McFeeley PJ, Oliver JM, Lipscomb MF. Immunohistochemical detection of human basophils in postmortem cases of fatal asthma. Am J Respir Crit Care Med 2001; 164: 1053-8.
Carroll NG, Perry S, Karkhanis A, Harji S, Butt J, James AL et al. The airway longitudinal elastic fiber network and mucosal folding in patients with asthma. Am J Respir Crit Care Med 2000; 161: 244-8.
Coskun S, Yuksel H, Tikiz H, Danahaliloglu S. Standard dose of inhaled albuterol significantly increases QT dispersion compared to low dose of albuterol plus ipratropium bromide therapy in moderate to severe acute asthma attacks in children. Pediatr Int 2001; 43: 631-6.
Abramson MJ, Bailey MJ, Couper FJ, Driver JS, Drummer OH, Forbes AB et al; Victorian Asthma Mortality Study Group. Are asthma medications and management related to deaths from asthma?. Am J Respir Crit Care Med 2001; 163: 12-8.
Turner MO, Noertjojo K, Vedal S, Bai T, Crump S, Fitzgerald JM. Risk factors for near-fatal asthma. A case-control study in hospitalized patients with asthma. Am J Respir Crit Care Med 1998; 157(6 Pt 1): 1804-9.
Champ CS, Byard RW. Sudden death in asthma in childhood. Forensic Sci Int 1994; 66: 117-27.
Burggraaf J, Westendorp RG, Veen JC, Schoemaker RC, Sterk PJ, Cohen AF et al. Cardiovascular side effects of inhaled salbutamol in hypoxic asthmatic patients. Thorax 2001; 56: 567-9.
Brown DL, Wetli CV, Davis JH. Sudden unexpected death from primary pulmonary hypertension. J Forensic Sci 1981; 26: 381-6.
Ackermann DM, Edwards WD. Sudden death as the initial manifestation of primary pulmonary hypertension. Report of four cases. Am J Forensic Med Pathol 1987; 8: 97-102.
Bolster MA, Hogan J, Bredin CP. Pulmonary vascular occlusive disease presenting as sudden death. Med Sci Law 1990; 30: 26-8.
Robalino BD, Moodie DS. Primary pulmonary hypertension, then and now: 28 years of experience. Cleve Clin J Med 1992; 59: 411-7.
Farnie DE, Storrow A, Whitley H. Syncope in a 2-year-old: ED presentation of primary pulmonary hypertension. Ann Emerg Med 1997; 30: 337-42.
Sudden cardiac death. En: Virmani R, Atkinson JB, Burke A, Farb A, editors. Cardiovascular Pathology. 2.ª ed. Philadelphia: WB Saunders; 2001. p. 340-85.
Graham MA, Hutchins GM. Forensic pathology. Pulmonary disease. Clin Lab Med 1998; 18: 241-62.
Patrat JF, Jondeau G, Dubourg O, Lacombe P, Rigaud M, Bourdarias JP, Gandjbakhch I. Left main coronary artery compression during primary pulmonary hypertension. Chest 1997; 112: 842-3.
Maitre B, Habibi A, Roudot-Thoraval F, Bachir D, Belghiti DD, Galacteros F et al. Acute chest syndrome in adults with sickle cell disease. Chest 2000; 117: 1386-92.
Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med 2000; 342: 1855-65.
Hiss J, Kahana T, Kugel C. Beaten to death: why do they die? J Trauma 1996; 40: 27-30.
Mudd KL, Hunt A, Matherly RC, Goldsmith LJ, Campbell FR, Nichols GR, Rink RD. Analysis of pulmonary fat embolism in blunt force fatalities. J Trauma 2000; 48: 711-15.
Knight B. Complications of injury. En: Knight B, editor. Forensic Pathology. 2.ª ed. London: ARNOLD; 1996. p. 333-45.
Clark SL, Hankins GD, Dudley DA, Dildy GA, Porter TF. Amniotic fluid embolism: analysis of the national registry. Am J Obstet Gynecol 1995; 172: 1158-67.
Kent KJ, Cooper BC, Thomas KW, Zlatnik FJ. Presumed antepartum amniotic fluid embolism. Obstet Gynecol 2003; 102: 493-5.
Cheung AN, Luk SC. The importance of extensive sampling and examination of cervix in suspected cases of amniotic fluid embolism. Arch Gynecol Obstet 1994; 255: 101-5.
Kobayashi H, Ooi H, Hayakawa H, Arai T, Matsuda Y, Gotoh K, Tarao T et al. Histological diagnosis of amniotic fluid embolism by monoclonal antibody TKH-2 that recognizes NeuAc alpha 2-6GalNAc epitope. Hum Pathol 1997; 28: 428-33.
Lunetta P, Penttila A. Inmunohistochemical identification of syncytiotrophoblastic cells and megakaryocytes in pulmonary vessels in a fatal case of amniotic fluid embolism. Int J Legal Med 1996; 108: 210-4.
Ishiyama I, Mukaida M, Komuro E, Keil W. Analysis of a case of generalized amniotic fluid embolism by demonstrating the fetal isoantigen (A blood type) in maternal tissues of B blood type, using immunoperoxidase staining. Am J Clin Pathol 1986; 85: 239-41.
Clark SL. New concepts of amniotic fluid embolism: a review. Obstet Gynecol Surv 1990; 45: 360-8.
Clark SL, Hankins GD, Dudley DA, Dildy GA, Porter TF. Amniotic fluid embolism: analysis of the national registry. Am J Obstet Gynecol 1995; 172(4 Pt 1): 1158-67.
Benson MD. Nonfatal amniotic fluid embolism. Three possible cases and a new clinical definition. Arch Fam Med 1993; 2: 989-94.
Benson MD, Kobayashi H, Silver RK, Oi H, Greenberger PA, Terao T. Immunologic studies in presumed amniotic fluid embolism. Obstet Gynecol 2001; 97: 510-4.
Fineschi V, Gambassi R, Gherardi M, Turillazzi E. The diagnosis of amniotic fluid embolism: an immunohistochemical study for the quantification of pulmonary mast cell tryptase. Int J Legal Med 1998; 111: 238-43.
Nishio H, Matsui K, Miyazaki T, Tamura A, Iwata M, Suzuki K. A fatal case of amniotic fluid embolism with elevation of serum mast cell tryptase. Forensic Sci Int 2002; 126: 53-6.
Rainio J, Penttilä A. Amniotic fluid embolism as cause of death in a car accident-a case report. Forensic Sci Int 2003; 137: 231-4.
Oliver J, Lyons TJ, Harle R. The role of computed tomography in the diagnosis of arterial gas embolism in fatal diving accidents in Tasmania. Australas Radiol 1999; 43: 37-40.
Thali MJ, Schweitzer W, Yen K, Vock P, Ozdoba C, Spielvogel E et al. New horizons in forensic radiology: the 60-second digital autopsy-full-body examination of a gunshot victim by multislice computed tomography. Am J Forensic Med Pathol 2003; 24: 22-7.
Bajanowski T, West A, Brinkmann B. Proof of fatal air embolism. Int J Legal Med 1998; 111: 208-11.
Start RD, Cross SS. Acp. Best practice no 155. Pathological investigation of deaths following surgery, anaesthesia, and medical procedures. J Clin Pathol 1999; 52: 640-52.
Ritz-Timme S, Eckelt N, Schmidtke E, Thomsen H. Genesis and diagnostic value of leukocyte and platelet accumulations around «air bubbles» in blood after venous air embolism. Int J Legal Med 1998; 111: 22-6.
Silver MM, Silver MD. Examination of the heart and of cardiovascular specimens in surgical pathology. En: Silver MD, Gotlieb AI, Schoen FJ, editors. Cardiovacular Pathology. New York: Churchill Livingstone; 2001. p. 1-29.
Tattersall RB, Gill GV. Unexplained deaths of type 1 diabetic patients. Diabet Med 1991; 8: 49-58.
Campbell I. Dead in bed syndrome: a new manifestation of nocturnal hypoglycaemia?. Diabet Med 199; 8: 3-4.
Borch-Johnson K, Helweg-Larsen K. Sudden death and human insulin: is there a link? Diabet Med 1993; 10: 255-9.
Thordarson H Sovik O. Dead in bed syndrome in young diabetic patients in Norway. Diabet Med 1995; 12: 782-7.
Sartor G, Dahlquist G. Short-term mortality ni childhood onset insulin-dependent diabetes mellitus: a high frequency of unexpected death. Diabet Med 1995; 12: 607-11.
Edge JA, Ford-Adams ME, Dunger DB. Causes of death in children with insulin dependent diabetes 1990-96. Arch Dis Child 1999; 81: 318-23.
Sovik O, Thordarson H. Dead-in-bed syndrome in young diabetic patients. Diabetes Care 1999; 22 Suppl 2: B40-2.
Osuna E, Garcia-Villora A, Perez-Carceles M, Conejero J, Maria Abenza J, Martinez P et al. Glucose and lactate in vitreous humor compared with the determination of fructosamine for the postmortem diagnosis of diabetes mellitus. Am J Forensic Med Pathol 2001; 22: 244-9.
Madea B, Kreuser C, Banaschak S. Postmortem biochemical examination of synovial fluid—a preliminary study. Forensic Sci Int 2001; 118: 29-35.
Winecker RE, Hammett-Stabler CA, Chapman JF, Ropero-Miller JD. HbA1c as a postmortem tool to identify glycemic control. J Forensic Sci 2002; 47: 1373-9.
Weston PJ, Gill GV. Is undetected autonomic dysfunction responsible for sudden death in Type 1 diabetes mellitus? The «dead in bed» syndrome revisited. Diabet Med 1999; 16: 626-31.
McNally PG, Lawrence IG, Panerai RB, Weston PJ, Thurston H. Sudden death in type 1 diabetes. Diabetes Obes Metab 1999; 1: 151-8.
Weston PJ, James MA, Panerai RB, McNally PG, Potter JF, Thurston H. Evidence of defective cardiovascular regulation in insulin-dependent diabetic patients without clinical autonomic dysfunction. Diabetes Res Clin Pract 1998; 42: 141-8.
Harris ND, Heller SR. Sudden death in young patients with Type 1 diabetes: a consequence of disease, treatment or both?. Diabet Med 1999; 16: 623-5.
Heller SR. Abnormalities of the electrocardiogram during hypoglycaemia: the cause of the dead in bed syndrome? Int J Clin Pract Suppl 2002; (129): 27-32.
Robinson RT, Harris ND, Ireland RH, Lindholm A, Heller SR. Comparative effect of human soluble insulin and insulin aspart upon hypoglycaemia-induced alterations in cardiac repolarization. Br J Clin Pharmacol 2003; 55: 246-51.
Robinson RT, Harris ND, Ireland RH, Lee S, Newman C, Heller SR. Mechanisms of abnormal cardiac repolarization during insulin-induced hypoglycemia. Diabetes 2003; 52: 1469-74.
LeCount ER, Singer HA. Fat replacement of the glycogen in the liver as a cause of death. Arch Pathol Lab Med 2001; 125: 15-20.
Chejfec G. Fat replacement of the glycogen in the liver as a cause of death: seventy-five years later. Arch Pathol Lab Med 2001; 125: 21-4.
Iten PX, Meier M. Beta-hydroxybutyric acid-an indicator for an alcoholic ketoacidosis as cause of death in deceased alcohol abusers. J Forensic Sci 2000; 45: 624-32.
Kadis P, Balazic J, Ferlan-Marolt V. Alcoholic ketoacidiosis: a cause of sudden death of chronic alcoholics. Forensic Sci Int 1999;103: S53-9.
Brinkmann B, Fechner G, Karger B, DuChesne A. Ketoacidosis and lactic acidosis—frequent causes of death in chronic alcoholics? Int J Legal Med 1998; 111: 115-9.
Pounder DJ, Stevenson RJ, Taylor KK. Alcoholic ketoacidosis at autopsy. J Forensic Sci 1998; 43: 812-6.
Day CP, James OF, Butler TJ, Campbell RW. QT prolongation and sudden cardiac death in patients with alcoholic liver disease. Lancet 1993; 341(8858): 1423-8.
Stratton SJ, Rogers C, Brickett K, Gruzinski G. Factors associated with sudden death of individuals requiring restraint for excited delirium. Am J Emerg Med 2001; 19: 187-91.
Britton A, McKee M. The relation between alcohol and cardiovascular disease in Eastern Europe: explaining the paradox. J Epidemiol Community Health 2000; 54: 328-32.
McKee M, Britton A. The positive relationship between alcohol and heart disease in eastern Europe: potential physiological mechanisms. J R Soc Med 1998; 91: 402-7.
![]()