
Vol. 37, n.º 1, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 1, 2004 |
Beatriz Aguilera Tapia, M.ª Paz Suárez Mier
Instituto Nacional de Toxicología. Sección de Histopatología. Madrid. b.aguilera@mju.es
RESUMEN
Se revisa el concepto de muerte súbita cardiaca y de muerte súbita arrítmica con corazón estructuralmente normal, así como la metódica de estudio recomendada para poder llegar a este diagnóstico. Se describen las entidades conocidas hasta el momento y que se deben a alteraciones en los canales iónicos cardiacos como son el síndrome de QT largo, el síndrome de Brugada y la Taquicardia Ventricular Polimórfica Catecolaminérgica. Se destaca la labor de prevención que puede hacer el patólogo al tratarse de síndromes que pueden ser familiares y que pueden debutar con muerte súbita, así como la necesidad de incorporar el análisis molecular al estudio autópsico para establecer el diagnóstico y dar el oportuno consejo genético a la familia del fallecido.
Palabras clave: Síndrome de muerte súbita inexplicada, síndrome QT largo, síndrome de Brugada, canalopatías, taquicardia ventricular.
SUMMARY
We review the concept of sudden cardiac death and of arrhythmic sudden cardiac death occurring with structurally normal heart. We describe the syndromes associated to cardiac ionic diseases (channelopathies) such as long QT syndrome, Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. We remark the preventing role that forensic pathologist have in these cases and the importance of molecular analysis for establishing the diagnoses and giving, if necessary, a genetic counsel to deceased families.
Key words: Sudden unexplained death syndrome, long QT syndrome, Brugada syndrome, ion channelopathies, ventricular tachycardia.
«Mors sine materia»
INTRODUCCIÓN
El estudio de los casos de muerte súbita, debido a lo inesperadas que son, o por las circunstancias que pueden ocurrir simulando una muerte violenta, accidental o una intoxicación, caen dentro del ámbito de acción de la medicina forense, constituyendo un porcentaje importante de la actividad tanatológica de los Institutos de Medicina Legal. La autopsia médico-legal de estos casos tiene como objetivo establecer la causa de la muerte.
La mayoría de las muertes súbitas son de origen cardiaco. La muerte súbita cardiaca (MSC) se define como aquella muerte natural, inesperada en el tiempo, que viene precedida por la pérdida brusca de conciencia dentro de, como máximo, la hora que sigue al comienzo de los síntomas agudos, en un sujeto con cardiopatía de base conocida o desconocida. Con gran frecuencia son muertes fulminantes y afectan a personas que se consideran sanas, o al menos carecen de síntomas, y ocurren mientras desarrollan su actividad habitual (en el trabajo, durante el sueño, practicando un deporte o conduciendo). En un porcentaje importante de casos la muerte súbita es la primera manifestación de la enfermedad cardiaca.
La cardiopatía isquémica, la hipertrofia cardiaca hipertensiva, las miocardiopatías y la estenosis aórtica son las causas más frecuentes de MSC.
La cardiopatía isquémica, que en el 99% de los casos tiene como base una ateroesclerosis coronaria, es la primera causa de muerte súbita en el mundo desarrollado. Es más frecuente que ocurra entre los 45 y 75 años de edad, ya que la población mayor tiende a fallecer por insuficiencia cardiaca. En España, la patología coronaria ateroesclerótica es la primera causa de muerte súbita a partir de los 30 años de edad, según un estudio epidemiológico hecho en menores de 36 años en Vizcaya (1), siendo la muerte súbita la forma de debutar de esta enfermedad que, en esta revisión, sólo afectaba a varones (2).
Sin embargo, existe un porcentaje de casos en los que un examen postmortem tradicional completo no identifica la causa del fallecimiento. Si se ha excluido una causa extracardiaca, estas muertes deben ser catalogadas como cardiacas, considerando que la parada cardiaca es el mecanismo que determina la muerte. Es lo que se ha denominado muerte súbita cardiaca arrítmica en individuos con corazón estructuralmente normal (3).
Es por todos conocido el síndrome de la muerte súbita del lactante, que ocurre dentro del primer año de edad. Sin embargo, es más desconocido que un número importante de las muertes súbitas que ocurren en niños, adolescentes y adultos jóvenes dan como resultado una autopsia en blanco, es decir, sin hallazgos patológicos que expliquen el fallecimiento inesperado de un joven sano que ha desarrollado su actividad habitual hasta el momento del deceso.
El porcentaje de autopsias en las que no se determina la causa de la muerte varía en los diversos estudios dependiendo del grupo de edad estudiado, del método de estudio y de los criterios diagnósticos. Por ejemplo, en las series de MSC publicadas por los patólogos de la AFIP, no encuentran hallazgos patológicos en el grupo de edad entre los 14-20, en el 30% de los casos, cifra que se reduce al 21% en el grupo con edad entre 21 y 30 años, y al 9% entre los 31 y 40 años (4). En el estudio de muerte súbita en jóvenes de Vizcaya, no se encontró patología estructural en un 18% de los casos de muerte súbita (1). Estos mismos autores estudian la casuística de muertes súbitas inexplicadas entre 1-35 años y lo desglosan por grupos de edad. Representan el 28,6% en el grupo entre 1-14 años, el 20,8% entre 15-29 años y el 4% entre los 30-35 años, obteniendo una tasa de mortalidad de muertes súbitas inexplicadas de 0,43 x 100.000 hab/año (5).
Por lo tanto, a medida que la MSC ocurre en personas de mayor edad, es más frecuente poder determinar la patología que la ha causado, pero entre los más jóvenes, aunque la MSC es muy infrecuente, en un alto porcentaje de casos la autopsia clásica no permite determinar la patología que ha producido el fallecimiento.
Parte de estas muertes, en principio inexplicadas, pueden corresponder a casos en los que una investigación de las circunstancias en las que se produjeron, y de los antecedentes, pueden ayudar a establecer su origen. Por ejemplo, muertes por golpe en el pecho en ausencia de lesión traumática cardiaca (commotio cordis), alteraciones electrolíticas asociadas a altas temperaturas, antecedentes de síndrome de preexcitación como el Wolff-Parkinson-White, epilepsia, etc.
REQUISITOS PARA HABLAR DE AUTOPSIA BLANCA
Para catalogar una autopsia como «blanca» y, por lo tanto, el mecanismo de la muerte como inexplicado, se tienen que cumplir una serie de requisitos:
1. Conocer las circunstancias de la muerte y el historial clínico personal y familiar. En casos de muertes no presenciadas, como las que ocurren durante el sueño, puede dar información el examinar el lugar del fallecimiento buscando substancias tóxicas, temperatura ambiente, etc.
2. Realizar una autopsia completa, con estudio microscópico de todos los órganos, que incluya el estudio del sistema de conducción cardiaco.
3. Análisis químico-toxicológico negativo.
4. Niveles de electrolitos y glucosa en el humor vítreo dentro de lo normal.
En las autopsias forenses se realiza análisis bioquímico del humor vítreo porque sus valores son equivalentes a los sanguíneos, y la muestra no sufre las precoces modificaciones postmortem de la sangre y el suero. Permite, de esta manera, determinar la existencia de una hiperglicemia y desórdenes hidro-electrolíticos.
Se ha propuesto el término de síndrome de muerte súbita inexplicada para aquellos casos en que no hay hallazgos patológicos en la autopsia, ni antecedentes patológicos o electrocardiográficos previos a la muerte (6). Sin embargo, muchas de ellas pueden encuadrarse en un grupo heterogéneo de arritmias familiares, que también pueden debutar con muerte súbita, y en las que los avances en patología molecular se están produciendo a gran velocidad. Los cuadros clínicos más importantes en este campo son el síndrome del QT largo, el síndrome de Brugada y la taquicardia ventricular familiar catecolaminérgica (7). Para comprender estos síndromes hay que recordar la electrofisiología cardiaca. La activación de las células cardiacas está regulada por las proteínas de los canales iónicos, que controlan el flujo de los iones de sodio, potasio y calcio a través de la membrana del miocito y, por lo tanto, son los responsables de la formación del potencial de acción transmembrana de la fibra miocárdica, que es diferente para cada tipo de célula muscular cardiaca (fig. 1). Este potencial de acción tiene 5 fases (8) (fig. 2):
Fase 0: es la fase de despolarización rápida, donde la entrada de iones Na+ a través de los canales de Na+ hace cambiar el potencial de –90 mv a +20 mv.
Fase 1: de repolarización temprana, por la entrada de iones Ca a través de sus correspondientes canales.
Fase 2: tiene forma de meseta, se produce por la salida lenta de los iones K+ hacia el exterior de la célula a través de los canales (IKr).
Fase 3: de repolarización rápida tardía, donde el paso de los iones K+ está muy facilitado a través de otros canales de este ión (IKS).
Fase 4: de reposo.
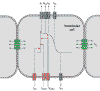
Fig. 1. Canales
iónicos que determinan el potencial de acción transmembrana.
![]()
Fig. 2. Fases del potencial de acción
transmembrana.
En los últimos años se ha avanzado enormemente en el estudio de la arquitectura de los canales iónicos, de sus locus cromosómicos y de las mutaciones que pueden aparecer, que son las responsables de las enfermedades que pasamos a describir.
SÍNDROME DEL QT LARGO
El síndrome del QT largo es una enfermedad de la repolarización cardiaca, que se identifica por una prolongación del intervalo QT en el ECG y que clínicamente se caracteriza por la aparición de arritmias ventriculares malignas, episodios de síncope y muerte súbita (7). Comprende un grupo de enfermedades cardiovasculares arritmogénicas, genéticamente determinadas, que resultan de la mutación de los genes que codifican los canales que regulan las corrientes de sodio y potasio y, por una mutación de un gen citoesquelético (Ankirina B), que puede afectar la cinética del canal de sodio (8).
Hasta el momento se han identificado 7 genes que determinan la prolongación del intervalo QT, los cuales se identifican por la sigla LQT y el número que refleja el orden en que han sido descubiertos, y por el nombre del gen mutado (tabla 1). Actualmente se ha demostrado que estos 7 genes mutados representan sólo el 50% de los casos de QT largo diagnosticados. De los casos conocidos, el 43% tiene mutado el LQT1; el 45%, el LQT 2 y el 7%, el LQT3 (9). La figura 2, además de representar las fases del potencial de acción, demuestra en qué fase actúan los genes mutados en las formas más frecuentes de síndrome QT conocidas (LQT1-3 y 5).

En esta patología de los canales iónicos, la localización de la alteración genética o genotipo, determina la expresión clínica o fenotipo. Al igual que en otras enfermedades genéticas, la penetrancia es variable. Esta variabilidad en la expresión en individuos con una misma mutación, probablemente se deba a la influencia de factores ambientales y/o a la coexistencia de otras mutaciones.
El alargamiento del intervalo QT puede ser asintomático o manifestarse por síncope, convulsiones y muerte súbita, que son secundarios a la aparición de una taquicardia ventricular polimórfica, con un aspecto peculiar en el trazado electrocardiográfico conocida como «Torsades de pointes», porque se curvan las puntas en el trazado electrocardiográfico (10) (fig. 3). El dato diagnóstico fundamental es la presencia de un intervalo QT corregido alargado (QTc = QT/[RR]1/2) en el ECG con valor superior a 0,46 segundos1/2. El QT largo puede ser hereditario, esporádico o adquirido. Este último, relacionado con medicamentos, alteraciones electrolíticas y otras enfermedades (tabla 2). En todas sus formas, las manifestaciones clínicas son semejantes (10).

Fig. 3. ECG típico de QT largo y
taquicardia vertricular conocida como "Torsades de pointes".

Se ha sugerido que un tercio de las muertes súbitas inexplicadas pueden ser atribuibles al síndrome del QT largo (11). También se ha asociado el síndrome del QT largo al síndrome de muerte súbita del lactante (12), planteándose que esta alteración del ritmo cardíaco podría ser la expresión de alteraciones en el desarrollo del sistema nervioso simpático, que no finaliza hasta los 6 meses de vida. Un aumento súbito en la actividad simpática podría desencadenar arritmias letales en estos corazones eléctricamente inestables.
Hasta el momento se han descrito tres síndromes con QT largo:
El síndrome de Jervell-Lange-Nielsen fue descrito en 1957, en una familia noruega donde 4 de los 6 hijos presentaban QT largo, sordera y síncope recurrente. Tres de ellos fallecieron súbitamente. Es muy infrecuente (1-6 casos/millón habitantes), y se transmite de forma autosómica recesiva (10).
El síndrome de Romano Ward se describió a principio de los años 60, se hereda con carácter autosómico dominante, de manera que teóricamente todos los portadores del gen tendrán manifestaciones clínicas, y la posibilidad de transmitir el defecto genético a su descendencia será del 50% (13). Sin embargo se ha visto que la penetrancia es baja y que puede haber portadores asintomáticos. La incidencia estimada de este síndrome es de 1/10.000 personas (8). Además de las manifestaciones clínicas ya mencionadas (síncope, convulsiones y muerte súbita) también puede asociarse a diabetes mellitus, asma y sindactilia (10).
El síndrome de Andersen es extremadamente infrecuente, pero es único entre las patologías de los canales iónicos, ya que se manifiesta por alteraciones del funcionamiento del músculo cardiaco y esquelético, coexistiendo la prolongación del QT con parálisis periódica (14).
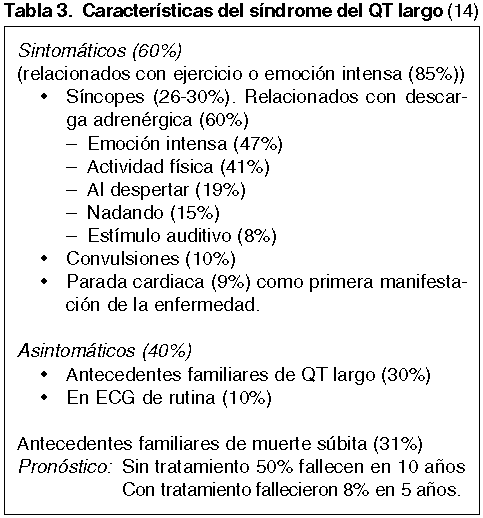
En la tabla 3 se resumen las características del síndrome QT largo según un estudio internacional de 287 pacientes pediátricos (15). Los desencadenantes auditivos observados en algunos casos fueron el ruido del despertador y el ruido del teléfono.
La determinación de la mutación que presenta un paciente afecto de síndrome de QT largo es importante porque se ha visto cierta correlación entre el genotipo (mutación) y el fenotipo (expresividad clínica). Así, las mutaciones del tipo LQT1 y LQT2 se asocian a síntomas precoces (sobre todo síncopes), pero el riesgo de muerte súbita es bajo. Por el contrario, aquellos con LQT3 muestran pocos síntomas, pero tienen mayor propensión a la muerte súbita (12). Además, en los LQT1 y LQT2 los síntomas están asociados al estrés y al ejercicio, incluyendo los estímulos sonoros, mientras que en los LQT3 los síntomas están asociados al sueño y al reposo (12,16). Los pacientes con LQT3 que presentan la mutación en el canal del Na+ podrían ser tratados con fármacos bloqueantes del sodio como la mexiletina. En los otros casos se aconsejan los ß-bloqueantes o los desfibriladores.
Implicaciones forenses en el síndrome de QT largo
Como ya se ha comentado y reflejado en la tabla 2, existen muchos fármacos que, de forma adquirida, pueden prolongar el intervalo QT y terminar en torsades de pointes. Se ha visto que algunos fármacos antiarrítmicos tienen este efecto, porque bloquean el canal de potasio codificado por el gen HERG, pero, el hecho de que solamente algunas personas tengan riesgo de presentar QT largo adquirido, sugiere una predisposición genética hacia una respuesta excesiva a los medicamentos (16). Makita et al. han demostrado esta susceptibilidad a desarrollar arritmias letales en un caso que presentaba mutación subclínica del SCN5A (17).
Algunos casos de muerte súbita ocurren en pacientes tratados con antiarrítmicos o antidepresivos, en los que en la autopsia no encontramos patología de suficiente importancia como para explicar la muerte, y los niveles de estos fármacos en sangre están en el rango terapéutico. En estos casos, debemos considerar la posibilidad de una muerte arrítmica por un QT largo adquirido en pacientes genéticamente predispuestos al efecto adverso de estos medicamentos, como hemos comentado en el párrafo anterior.
Ackerman et al. han publicado un artículo de gran interés. Presentan el caso de una muerte súbita inexplicada en un varón de 17 años. La madre y la abuela tenían antecedentes de síncopes en diferentes momentos de su vida. Se hizo una evaluación clínica de los familiares, que determinó el diagnóstico de síndrome de QT largo en la madre, tras un test de provocación con epinefrina. Esta respuesta al test de la epinefrina les condujo a buscar una mutación en el gen KVLQT1 (KCNQ1), que interviene en la fase 3 del potencial de acción (responsable del síndrome QT largo tipo 1) en las muestras de tejido del fallecido, identificándose una mutación en este gen, lo que permitió establecer la causa de la muerte y realizar tratamiento preventivo en un hermano de 13 años (6).
También resulta muy interesante el estudio que realizaron los mismos autores, sobre muestras congeladas de miocardio procedentes de la autopsia de una mujer de 19 años, que falleció a los 12 días de ser rescatada del fondo de una piscina de escasa profundidad. Aunque respondió a las medidas de resucitación con desfibrilador, falleció por encefalopatía anóxica. Durante su estancia hospitalaria, presentó QT largo e inestabilidad eléctrica en el ECG. El estudio genético demostró una nueva mutación en el gen KVLQT1, que también se detectó en su madre, abuela y una hermana, lo que permitió el consejo genético (riesgo del 50% de transmitir la mutación a su descendencia) y asesoramiento sobre el riesgo de muerte súbita asociada a la natación y otros ejercicios físicos. Así mismo se aconsejó tratamiento protector con ß bloqueantes (18).
Lunetta et al. enfatizan la trascendencia que pueden tener los estudios de biología molecular en la medicina forense (19). Presentan el caso de una mujer de 44 años, en tratamiento por depresión, a la que encuentran desnuda, muerta en la bañera. Debido a que esta mujer tenía antecedentes de varios intentos autolíticos, aunque no había dejado nota suicida, el caso se clasificó como muerte no natural, suicida. Sin embargo, la revisión del historial clínico demostró, en un ECG tomado tres meses antes de su fallecimiento, la existencia de un intervalo QT corregido aumentado. Al efectuar el estudio molecular, se encontró mutación del gen KCNH2. Esto, en medicina forense tiene una gran trascendencia, porque obliga a plantear que la causa del fallecimiento pudiera ser natural y, por lo tanto, cambia la etiología médico-legal de la muerte. Este hallazgo puede resultar fundamental para los familiares, tanto por el consejo médico que deben recibir, como por asuntos económicos si hay seguros de vida (no se pagan en casos de suicidios).
SÍNDROME DE BRUGADA
El síndrome de Brugada se define en base a criterios clínicos y electrocardiográficos. Se trata de un síndrome caracterizado por episodios de síncope y muerte súbita, en corazones estructuralmente normales, en donde el ECG presenta patrón de bloqueo de rama derecha y elevación del segmento ST en las derivaciones precordiales V1 a V3 (fig. 4) (20-22).
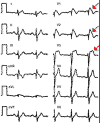
Fig. 4. ECG típico del Síndrome de
Brugada (tomado de www.crtia.be/about/picturepage.htm).
Fue descrito por Pedro y Josep Brugada, en 1992, a partir del estudio de 8 pacientes que habían presentado varios episodios de muerte súbita abortada (23). En los años 80, el Centro de Control de Enfermedades (CDC) de Atlanta alertó sobre el gran número de jóvenes refugiados procedentes del sureste asiático (muchos de Vietnam) y aparentemente sanos, que fallecían súbitamente durante la noche, y sin alteración estructural cardiaca. Por otra parte, también en países del este asiático (Tailandia, Filipinas y Japón) existe un elevado número de muertes súbitas inexplicadas en varones jóvenes (26-38/100.000 varones/año). En 1997 se descubrió que muchos de estos casos tenían alteraciones electrocardiográficas idénticas a las del síndrome de Brugada (24).
Desconocemos la incidencia de este síndrome en España. En Japón, se han hecho varios estudios prospectivos que han demostrado una incidencia de 0,05% de los adultos con la alteración típica en el ECG; 0,6% según otro estudio; 0,0006% en niños y adolescentes. Se estima que del 4-12% de las muertes súbitas e inesperadas, sobre todo en jóvenes, pueden ser debidas a este síndrome (21). En Bélgica y España se considera que del 40-60% de los casos de fibrilación ventricular idiopática pueden ser debidos al síndrome de Brugada (25).
El síndrome de Brugada es una enfermedad que se hereda con patrón autosómico dominante. En el 60% de los pacientes diagnosticados existen antecedentes de muerte súbita en la familia, se encuentran familiares asintomáticos con las mismas características electrocardiográficas o se producen nuevas muertes súbitas en la familia mientras se está estudiando al paciente. El primer gen que se relacionó con el síndrome de Brugada fue publicado a principios de 1998, estudiando 6 familias y dos casos esporádicos. En tres familias se identificaron mutaciones del gen del canal del sodio SCN5A, el mismo gen responsable del LQT3, pero en otros puntos (25). La heterogeneidad del periodo refractario debido a las anomalías en los canales de sodio facilita el desarrollo de arritmias por reentrada (22). Puesto que en una familia no se encontró ninguna mutación en este gen, se especula en la heterogeneidad genética de este síndrome (25). Posteriormente, Grant et al. han demostrado que este mismo gen SCN5A es responsable de alteraciones en la conducción, por lo tanto interviene en tres cuadros clínicos diferentes (26).
El síndrome completo se caracteriza por la aparición de episodios de taquicardia ventricular polimórfica rápida en pacientes con el ECG típico, que pueden causar síncope si terminan espontáneamente, o muerte súbita arrítmica cuando persisten y no son terminadas con desfibrilador. Se piensa que la acción moduladora del sistema nervioso autónomo puede intervenir para que evolucionen de una forma u otra. Algunos autores japoneses señalan que las muertes súbitas que se producen durante el sueño pueden ser desencadenadas por bradicardias (24).
Las arritmias pueden ser desencadenadas también por el alcohol y el stress (25) y también asociado a fiebre, quizás por la sensibilidad de los canales de sodio a la temperatura (27).
Hay pacientes asintomáticos donde el síndrome se descubre en un ECG casual o por muerte súbita de un familiar, y formas sintomáticas que debutan con síncope o fibrilación ventricular idiopática. El pronóstico es malo en ambas circunstancias, con una mortalidad del 40%. Un tercio de los sujetos diagnosticados de forma casual desarrollan un episodio de fibrilación ventricular en los dos años siguientes (23).
El diagnóstico no entraña dificultad cuando el ECG es típico. Sin embargo, hay casos en que las alteraciones son mínimas o incluso hay casos con ECG totalmente normales. Por ello, se administran antiarrítmicos, como la ajmalina, procainamida o flecainida, para desenmascarar las alteraciones ECG. Esto hay que tenerlo en cuenta para el estudio de pacientes con síncope de origen desconocido y también, en lo que nos concierne, para el estudio de familiares de víctimas de muerte súbita. El tratamiento de elección, y que da buenos resultados para la prevención de la muerte súbita, es la colocación de un desfibrilador implantable (23,27).
TAQUICARDIA VENTRICULAR POLIMÓRFICA CATECOLAMINÉRGICA
Se trata de una arritmia, caracterizada por taquicardia ventricular bidireccional inducida por el estrés físico y emocional, que afecta a niños y adolescentes, en ausencia de patología estructural y de prolongación del intervalo QT en el ECG, que puede degenerar en parada cardiaca y muerte súbita. Es una arritmia altamente reproducible y en un tercio de los casos existe historia familiar de muerte súbita precoz o de síncopes inducidos por el estrés o el ejercicio. Tiene una herencia autosómica dominante, con una tasa de mortalidad estimada a los 30 años del 30% (7). Electrocardiográficamente, es semejante a las arritmias asociadas a la hipercalcemia y a la toxicidad por digitálicos (28).
Una historia familiar característica es la publicada por Myrianthefs et al (29), donde describen el estudio de una familia motivado por el fallecimiento precoz de tres hermanos: dos hermanas a los 12 y 16 años mientras competían en natación y en una carrera, respectivamente, y del hermano de 19 años, que fallece súbitamente tras un accidente leve, conduciendo sin licencia un coche robado, en ausencia de lesiones traumáticas. En ninguno de los tres casos en la autopsia había lesiones estructurales que explicaran el fallecimiento súbito y presenciado de estos jóvenes. Estudiada por el cardiólogo la familia, encontró que la madre y una hermana de 19 años tenían antecedentes de síncopes, y ausencia de alteraciones al ECG de reposo o de anomalía estructural con el ecocardiograma. Sin embargo, al someterlas a un ECG de estrés, desarrollaron una taquicardia ventricular polimórfica.
Recientemente, se han identificado mutaciones en el gen hRyR2 que codifica el receptor de la rianodina cardiaca, cuyo locus está en el cromosoma 1 (1q42-q43) (28). El receptor de la rianodina juega un papel fundamental en la regulación de los flujos intracelulares de calcio y en la unión excitación-contracción, ya que el ión calcio se une a la Troponina C e inicia el proceso de contracción de la sarcómera (7,30). En una forma autosómica recesiva se han descubierto mutaciones en el locus 1p13-21 (31).
CONCLUSIÓN
En el momento actual, cierto número de casos de muerte súbita en niños, adolescentes y adultos jóvenes son informados como «Muerte súbita con corazón estructuralmente normal». Hecho este diagnóstico, es importante explicar a los familiares del fallecido que esta muerte puede deberse a síndromes que producen arritmias cardiacas, y que otros miembros de la familia pueden estar afectados y, por lo tanto, se les debe recomendar que consulten con un cardiólogo experto en arritmias para que estudien a los familiares directos. Es importante que el patólogo comprenda la angustia que esta indicación puede crear a la familia, que ya está abatida por la pérdida de su ser querido. Por ello, es fundamental que en todos los casos de muerte súbita se realice un estudio reglado del corazón, por la trascendencia del diagnóstico que se haga.
Confiamos que, en un futuro no lejano, las técnicas de biología molecular se aplicarán también a las autopsias, que se convertirán en autopsias moleculares (6), fundamental para el estudio de los casos de muertes súbitas inexplicadas, permitiendo llegar al diagnóstico molecular de la causa de muerte. Esto facilitaría enormemente el estudio del grupo familiar y el tratamiento adecuado de los miembros afectados.
Además, el desarrollo del diagnóstico molecular de estos casos, nos permitirá dar una explicación adecuada sobre la causa del fallecimiento. Actualmente, a estos familiares que probablemente están pasando por el trance más doloroso de su vida, les resulta muy difícil comprender que su ser querido, al que consideraban completamente normal y sano, haya fallecido en forma tan inesperada, y sin que se haya podido determinar la causa del deceso.
Citando a Brugada (32), debemos decir que «la medicina forense, al igual que todas las demás especialidades médicas, está obligada a adaptarse a los nuevos conocimientos y a las nuevas tecnologías, especialmente genéticas». Así podrá contribuir al estudio de las enfermedades, sin olvidar el importante rol preventivo que puede desarrollar, especialmente en estos casos de muertes inesperadas precoces, que son tan importantes y dolorosas para la familia, los amigos y para la sociedad.
BIBLIOGRAFÍA
Morentin B, Suarez-Mier MP, Audicana C, Aguilera B, Garamendi PM y Elexpe X. Incidencia y causas de muerte súbita en menores de 36 años. Med Clin (Barc) 2001; 116: 281-5.
Morentin B, Suarez-Mier MP, Aguilera B. Muerte súbita por enfermedad ateromatosa coronaria en jóvenes. Rev Esp Cardiol 2001; 54: 1167-74.
Thiene G, Basso C, Corrado D. Cardiovascular causes of sudden death. En: Silver M, Gotlieb A, Schoen F. Cardiovascular Pathology. Philadelphia: Churchill Livingstone; 2001. p. 326-74.
Virmani R, Burke A, Farb A y Atkinson J. Cardiovascular Pathology. 2.ª ed. Philadelphia: Saunders Co; 2001. p. 341-44.
Morentin B Suarez-Mier MP, Aguilera B. Sudden unexplained death among persons 1-35 years old. Forensic Sci Int 2003; 135: 213-17.
Ackerman MJ, Tester DJ, Driscoll DJ. Molecular autopsy of sudden unexplained death in the young. Am J Forensic Med Pathol 2001; 22: 105-11.
Roberts R, Brugada R. Genetics and Arrhytmias. Annu Rev Med 2003; 54: 257-67.
Moss AJ. Long QT Syndromes. JAMA 2003; 289: 2041-44.
Towbin JA, Wang Z, Li H. Genotype and severity of long QT syndrome. Drug Metab Dispos 2001; 29: 574- 9.
Ackerman MJ. The long QT syndrome: ion channel disease of the Heart. Mayo Clin Proc 1998; 73: 250-269.
Davies MJ. The investigation of sudden cardiac death. Histopathology 1999; 34: 93-8.
Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 1998; 338: 1709-14.
Brugada R. Bases genéticas de las arritmias. Rev Esp Cardiol 1998; 51: 274-85.
Tristani-Firouzi, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 2002: 110: 381-8.
Garson A Jr, Dick M 2nd, Fournier A, Gillette PC, Hamilton R, Kugler JD, van Hare GF 3rd, Vetter V, Vick GW 3rd. The long QT syndrome in children. An international study of 287 patients. Circulation 1993; 89: 1866-72.
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001; 103: 89-95.
Makita N, Horie M, Nakamura T, Ai t, Sasaki K, Yokoi H, et al. Drug-induced long QT syndrome associated with a subclinical SCN5A mutation. Circulation 2002; 106: 1269-74.
Ackerman MJ, Tester D, Porter CJ, Edwards WD. Molecular diagnosis of the inherited Long-QT syndrome in a woman who died after near-drowning. N Engl J Med 1999; 341: 1121-5.
Lunetta P, Levo A, Männikkö A, Penttilä A, Sajantila A. Death in bathtub revisited with molecular genetics: a victim with suicidal traits and a LQTS gene mutation. Forensic Sci Int 2002; 130: 122-4.
Brugada J. Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3. A marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998; 97: 457-60.
Brugada J, Brugada P, Brugada R. El síndrome de Brugada y las miocardiopatías derechas como causa de muerte súbita. Diferencias y similitudes. Rev Esp Cardiol 2000; 53: 275-85.
Gussak I, Antzelevitch C, Bjerregaard P, Towbin JA, Chaitman BR. The Brugada syndrome: clinical electrophysiologic and genetic aspects. J Am Coll Cardiol 1999; 33: 5-15.
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicentric report. J Am Coll Cardiol 1992; 20: 1391-96.
Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997; 97: 2595-600.
Chen Q, Kirsh GE, Zhang D, Brugada R, Brugada J, Brugada P et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998; 392: 293-6.
Grant AO, Carboni MP, Neplioueva V. Long QT syndrome, Brugada syndrome and conduction system disease are linked to a single sodium channel mutation. J Clin Invest 2002; 110: 1201-9.
González-Rebollo JM, Hernández Madrid A, García A, García de Castro A, Mejías A, Moro C. Fibrilación ventricular recurrente durante un proceso febril en un paciente con síndrome de Brugada. Rev Esp Cardiol 2000; 53: 755-7.
Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001; 103: 196-200.
Myriantheks M, Cariolou M, Eldar M, Minas M, Zambartas C. Exercise-Induced Ventricular Arrhythmias and sudden cardiac death in a Family. Chest 1997; 111: 1130-3.
Napolitano C, Priori S. Genetics of ventricular tachycardia. Curr Opin Cardiol 2002; 17: 222-8.
Lahat H, Eldar M, Levy-Nissenbaum E. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation 2001; 103: 2822-7.
Brugada J. Muerte súbita en jóvenes. Med Clin (Barc) 2001; 116: 294-5.
![]()