
Vol. 37, n.º 2, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 2, 2004 |
Juan F. García, Miguel A. Piris, Manuel M. Morente
Programa de Patología Molecular. Centro Nacional de Investigaciones Oncológicas.
RESUMEN
El objetivo de cualquier clasificación de linfomas es proporcionar una herramienta internacional de comunicación. La clasificación debe ser reproducible y clínicamente relevante, así como suficientemente flexible como para permitir la incorporación de nueva información. Por último, debe estar basada en criterios histopatológicos porque es el patólogo quien, en la mayoría de las ocasiones, hace el diagnóstico inicial. La actual clasificación de la OMS de las neoplasias linfoides representa un avance fundamental en nuestro conocimiento de estos tumores. Esta clasificación prioriza el estado de diferenciación de la célula tumoral, agrupando las neoplasias por criterios morfológicos, histológicos, inmunofenotípicos, citogenéticos, moleculares y clínicos. En referencia a los tumores de células B, la categoría de linfomas B de células pequeñas incluye la leucemia linfática crónica, linfoma de células del manto, linfoma folicular, linfoma linfoplasmacítico/inmunocitoma, leucemia de células peludas y el plasmocitoma/mieloma. La categoría de linfomas de células grandes difusos es heterogénea e incluye diferentes subtipos tumorales. Las características moleculares están bien definidas en linfomas B de células pequeñas, como translocaciones cromosómicas afectando los genes de las inmunoglobulinas y los genes Bcl-2 o Ciclina D1. En esta revisión se compendian las principales características necesarias para el diagnóstico y categorización de los linfomas de células B, así como la información relativa a las características biológicas y clínicas de estos linfomas.
SUMMARY
The aim of any lymphoma classification is to provide an international language allowing communication. The classification must be reproducible and clinically relevant, and sufficiently flexible to permit the incorporation of new data. Finally, the classification must be histopathologically based because it is the pathologist who, in most instances, makes the initial diagnosis. The current WHO classification of lymphoid neoplasms represents a major step forward in our understanding of these tumours. This classification highlights the stage of differentiation of the tumour cell, grouping neoplasms by morphology, histology, immunophenotypes, cytogenetic, molecular abnormalities and clinical data. With regard to B-cell tumors, the small cell B-cell lymphomas category consist of chronic lymphocytic leukaemia, mantle cell lymphoma, follicular lymphoma, lymphoplasmocytic lymphoma/immunocytoma, hairy cell leukaemia, as well as plasmacytoma. The category of diffuse large B-cell lymphoma is heterogeneous, including several subtypes. The molecular biological characteristics are, in general, well defined in small cell B-cell lymphomas, such as chromosomal translocations involving the immunoglobulin heavy chain genes and the Bcl-2 gene or Cyclin D1 gene.
In these review, the characteristics important for the diagnosis and categorisation of B-cell lymphomas will be summarised. Further, we present information regarding the molecular biological and clinical characteristics of these lymphomas.
La existencia de diferentes clasificaciones de linfomas ha constituido siempre una fuente de frustración y controversia para clínicos, patólogos e investigadores básicos, convirtiendo en muchas ocasiones el diagnóstico histopatológico de estos tumores en un ejercicio de imaginación. En 1994 la revista Blood publicó la clasificación REAL de Linfomas, propuesta por el International Lymphoma Study Group, un grupo de expertos de Europa, Estados Unidos y Asia. Esta clasificación está basada en información clínica, morfológica y fenotípica acumulada durante décadas y rápidamente ganó consenso. La aceptación de esta clasificación ha sido facilitada por diversos estudios demostrando su reproducibilidad y haciendo manifiesto su significado clínico. Cada tipo tumoral definido tiene una presentación clínica y respuesta al tratamiento diferenciada, con variables pronosticas independientes, y es candidato a terapias dirigidas específicamente.
Este proceso de consenso se ha prolongado después de la publicación de la clasificación REAL con la iniciativa de la OMS de publicar un Libro Azul sobre linfomas y leucemias. Las Sociedades Americana y Europea de Hematopatología, con participación adicional de un numeroso grupo de hematólogos y oncólogos de todo el mundo han llevado a cabo una tarea de reedición de las entidades descritas en la clasificación REAL dando así lugar a la clasificación de la OMS, publicada en otoño del año 2001. Esta nueva clasificación incluye también las leucemias no linfoides, extendiendo de esta forma el consenso a todo el campo de los tumores de origen hemopoyético.
En la definición de entidades se ha dado prioridad a la simplificación, evitando hacerla compleja y de menor valor practico: sólo se reconocen enfermedades con rasgos característicos, tanto morfológicos como clínicos. Sin embargo, variaciones de agresividad o grado histológico dentro de una misma enfermedad no son reconocidas como enfermedades diversas.
En el caso de los linfomas B, estas definiciones toman aun mayor valor. La clasificación (tabla I) presta mayor relevancia que en el pasado a la presencia de alteraciones moleculares características de enfermedades concretas (tabla II), y en cambio presta menor atención a variaciones citológicas sutiles, difícilmente reproducibles. La localización anatómica de los diversos procesos linfoproliferativos B también es un marcador relevante, que determina las características biológicas y clínicas de cada enfermedad (tabla III).

La integración de datos genéticos y moleculares ha permitido validar muchas de las entidades propuestas, y al mismo tiempo matizar algunos aspectos, reconociendo también el valor práctico y potencial de marcadores moleculares para prever la conducta clínica y respuesta al tratamiento de las neoplasias hematológicas. De hecho, en la actualidad existe una general aceptación de la necesidad absoluta de complementar los estudios morfológicos y fenotípicos con técnicas moleculares en el diagnóstico rutinario de los linfomas. El valor potencial de estos marcadores moleculares modificará a medio plazo nuestra forma de estudiar y tratar los linfomas y leucemias. Eventualmente, esta clasificación de la OMS será actualizada recogiendo las aportaciones y los acuerdos de la comunidad científica y los progresos que se generen en el futuro en diagnostico y tratamiento de linfomas y leucemias.
En nuestro medio, el 80% de los procesos linfoproliferativos corresponden a neoplasias de células B, siendo los más frecuentes la leucemia linfática crónica B (27%), mieloma/plasmocitoma (24%) y linfomas B difusos de células grandes (16%). En esta revisión se enumeran las principales características diagnósticas de las entidades reconocidas por la OMS, incluyendo aspectos morfológicos, fenotípicos y moleculares.
LINFOMA/LEUCEMIA LINFOBLASTICO B
Neoplasia de linfoblastos B, que puede cursar de forma exclusivamente leucémica o con formación de masas tumorales en ganglio linfático, hueso, piel u otras localizaciones extraganglionares.
Rasgos clínicos: La mayoría de los casos se dan en pacientes jóvenes (<20 años) y con presentación leucémica. De forma infrecuente, la enfermedad puede debutar como tumores sólidos, sin compromiso leucémico.
Morfología: Los linfoblastos tienen una morfología característica, con tamaño celular intermedio, núcleo redondo o escotado, cromatina fina, nucleolo poco evidente y escaso citoplasma (fig. 1).
![]()
Fig. 1: Linfoma linfoblástico B. Intensa
expresión de proteína TdT en la mayoría de los núcleos tumorales.
Inmunofenotipo y genotipo: Tdt+, CD19+, CD79a+, CD20-/+, CD10+/-, CD34+/-. La demostración de Tdt es crítica en el diagnóstico. Algunos casos pueden ser CD99+. Un rasgo frecuente es la expresión de marcadores inmunohistoquímicos aberrantes, como CD3 o CD43.
Además de reordenamiento monoclonal del gen de la cadena pesada de las inmunoglobulinas (IgH), se puede encontrar también reordenamiento en los genes del receptor de células T (TCR). Algunas translocaciones se asocian a cursos clínicos diferentes, y se recomienda incluir esta información en el informe del paciente, así:
Translocaciones afectando el gen MLL en 11q23 conllevan un mal pronóstico.
Hiperdiploidía y translocación t(12; 21) (TEL/AML1) se asocian a un pronóstico favorable.
Translocación t(9; 22) (BCR/ABL) se asocia a pronostico desfavorable.
Translocación t(1; 19) (E2A-PBx), asociada a pronóstico desfavorable.
Variantes: Existe un consenso generalizado acerca de que las formas que cursan como tumorales y las leucémicas son sustancialmente la misma enfermedad en diferentes fases o con tropismo por diferentes órganos. No obstante, para evitar confusiones se recomienda mantener la denominación de leucemia para aquellas caracterizadas por presentación leucémica.
La terminología FAB no parece actualmente adecuada, ya que muestra una pobre correlación con el fenotipo, genotipo o manifestaciones clínicas de la enfermedad.
LEUCEMIA LINFOCÍTICA CRÓNICA B (LLC-B) / LINFOMA DE LINFOCITOS PEQUEÑOS
Rasgos clínicos: Enfermedad indolente, no curable y usualmente leucémica, de adultos de más de 40 años con afectación constante de médula ósea y frecuente de ganglios linfáticos, bazo e hígado. Ocasional presencia discreta de componente M en suero.
Morfología: Citología compuesta por linfocitos pequeños, prolinfocitos y parainmunoblastos con formación de seudofolículos (fig. 2). Presencia de folículos linfoides atrapados.

Fig. 2: LLC. Patrón difuso con presencia
de centros de proliferación (pseudofolículos). La citología está compuesta por
linfocitos maduros pequeños, prolinfocitos y parainmunoblastos.
Inmunofenotipo y Genotipo: CD23, CD43, IgD, CD5 positivo. Débil expresión de CD20, fuerte expresión de proteína Bcl2. Ausencia de Ciclina D1, Bcl6 y CD10. No existe una alteración citogenética patognomónica de esta entidad. Sin embargo, algunas de las alteraciones que se pueden observar son particularmente útiles en rutina diagnóstica:
Trisomía 12: 1/3 de los casos, con morfología atípica y mayor agresividad clínica.
Deleciones de 13q14, distales al gen de retinoblastoma.
Deleciones de 11q22-23 en 20% de los casos, asociados con marcadas adenomegalias y supervivencia acortada.
Mutaciones de p53 (frecuentemente asociadas con deleción de 17p13) le confieren mayor agresividad clínica, con resistencia a la quimioterapia.
La ausencia de mutaciones somáticas del gen de las inmunoglobulinas, observada en un 50% de los pacientes, se correlaciona con un peor pronóstico. La ausencia de mutaciones somáticas, y la supervivencia más corta, frecuentemente se asocia adicionalmente con expresión de CD38.
Variantes: Ocasionalmente pueden verse formas no leucémicas, de presentación ganglionar, para las cuales se usa la denominación de linfoma de linfocitos pequeños. Para el diagnóstico se requiere la misma composición citológica y patrón arquitectural, ya que la denominación de linfoma de linfocitos pequeños es muy ambigua y no debe usarse en un sentido genérico. No existen datos que justifiquen reconocer esta variante como una enfermedad diferente.
En algunos casos las células neoplásicas tienen un tamaño pequeño, pero el contorno nuclear aparece irregular, con indentaciones única o múltiples. Un leve grado de diferenciación plasmocitoide no es infrecuente. Algunos de estos casos han sido denominados en el pasado como inmunocitomas linfoplasmocitoides, y algunos estudios muestran un pronóstico ligeramente más adverso para este grupo de casos.
La presencia de seudofolículos no es constante, y puede depender del grado de afectación del ganglio. Sin embargo, se trata de un rasgo morfológico absolutamente patognomónico de la enfermedad. Una presencia masiva de seudofolículos se suele asociar con formas más tumorales, pero no está claro que tengan un significado pronóstico desfavorable.
LEUCEMIA PROLINFOCÍTICA B (LPL-B)
Rasgos clínicos: típicamente se presenta como esplenomegalia marcada, sin linfadenopatía. Edad superior a 60 años. Linfocitosis muy alta en sangre periférica (con frecuencia >100x103/mm3). Frecuente anemia y trombocitopenia. Raro componente M. Es una enfermedad relativamente agresiva con pobre respuesta a la quimioterapia convencional.
Morfología: Patrón difuso, sin formación de seudofolículos. En médula ósea, infiltración difusa, sobre todo intertrabecular. En bazo, infiltración de pulpa blanca y roja, con nódulos grandes en la pulpa blanca. Los prolinfocitos se definen como células de mediano tamaño, doble que un linfocito pequeño, cromatina nuclear moderadamente condensada, nucleolo grande destacado central, relativamente baja ratio núcleo/citoplasma, contorno nuclear usualmente regular (fig. 3).
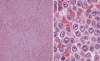
Fig. 3: Infiltración difusa del bazo por
LPL. Los prolinfocitos son células de tamaño intermedio, núcleo redondeado y
nucleolo central llamativo.
Inmunofenotipo y genotipo: Fuerte expresión de inmunoglobulina de superficie (sIg), IgM/D. FMC7+, CD79b+, CD23-, CD5-/+.
Es posible detectar translocaciones afectando 14q32, por lo que la definición de esta entidad muestra cierto grado de solapamiento con formas blastoides leucémicas de linfomas del manto. Actualmente se clasifican como linfoma de células del manto tumores con translocación t(11; 14). Existen frecuentes mutaciones de p53, que en algunas series alcanzan el 50% de casos.
LEUCEMIA DE CÉLULAS PELUDAS
Rasgos clínicos: Pacientes adultos con esplenomegalia, pancitopenia y usualmente linfocitosis tumoral discreta. Curso indolente, con buena respuesta a interferón, deoxicoformicina o 2-clorodeoxiadenosina.
Morfología: Proceso linfoproliferativo que afecta preferentemente a bazo y médula ósea. Las células tumorales en la sangre periférica tienen unas prolongaciones citoplasmáticas vellosas características, que se hacen evidentes en el frotis de sangre. La afectación medular intersticial y difusa es la regla, con fibrosis reticulínica. En el bazo la infiltración es fundamentalmente en la pulpa roja, generalizada, con formación de sinusoides revestidos por células neoplásicas (fig. 4).

Fig. 4: Leucemia de células peludas.
Expresión Inmunohistoquímica de DBA44.
Inmunofenotipo y genotipo: CD20+ CD5- CD10- CD23- CD11c+ CD25+ FMC7+ CD103+. En secciones parafinadas la inmunotinción con el anticuerpo DBA44 es útil para reconocer las células neoplásicas, especialmente en biopsias de médula ósea, en las que la afectación intersticial puede fácilmente pasar desapercibida. Algunos casos pueden mostrar expresión nuclear de Ciclina D1.
MIELOMA MÚLTIPLE
Neoplasia de células plasmáticas, con lesiones osteolíticas, plasmocitosis medular atípica y gammapatía monoclonal.
Rasgos clínicos: La mayoría de los pacientes tiene más de 50 años. La destrucción ósea produce lesiones osteolíticas, dolor óseo, fracturas patológicas, hipercalcemia y anemia. La proteinuria monoclonal puede conducir a fallo renal. La mayoría de los enfermos tiene un componente M sérico. Los criterios clásicos para el diagnóstico clínico de mieloma múltiple siguen siendo requisito indispensable.
Morfología : plasmocitosis superior al10% de las células nucleadas de la médula ósea (la densidad del infiltrado tiene valor pronóstico). Estas células plasmáticas aparecen además con una distribución irregular en la médula ósea, formando agregados de distribución perivascular. Citología variable incluyendo formas anaplásicas.
Inmunofenotipo y genotipo: Ig monoclonal citoplasmática, más frecuentemente IgG ó IgA. Puede verse sólo cadenas ligeras. La mayoría de los casos son CD20-, CD38+, CD79a+, CD138+, CD56+, CD45-/+. Otros marcadores inmunohistoquímicos son también útiles en el reconocimioento de las células plasmáticas: EMA, PAX5, MUM1.
Con frecuencia se detectan translocaciones de IgH con Ciclina D1 (es frecuente detectar expresión nuclear de Ciclina D1), c-myc, CDK6 o CiclinaD3.
Variantes morfológicas:
Plasmablástica, asociada con peor pronóstico.
Mieloma no secretor
Quiescente (smoldering)
Indolente
Leucemia de células plasmáticas
Mieloma osteoesclerótico (síndrome de POEMS)
PLASMOCITOMA
Neoplasia de células plasmáticas, limitada a tumefacción localizada ósea o extraesquelética (tracto respiratorio superior, tracto gastrointestinal y otros). Raro componente M sérico. Se reconoce como una forma localizada de presentación de mieloma, con morfología e inmunofenotipo superponibles. La demostración inmunohistoquímica en las biopsias de restricción de cadenas ligeras kappa o lambda, tiene un valor crítico en el diagnóstico diferencial.
LINFOMA LINFOPLASMACÍTICO/ INMUNOCITOMA
Tumor derivado de linfocitos B con marcada diferenciación plasmocitoide, frecuentemente asociado a síndrome clínico de macroglobulinema de Waldenstrom, aunque esto no es un requisito indispensable. Se trata de un diagnóstico de exclusión, ya que muchos procesos linfoproliferativos B de bajo grado pueden presentar rasgos plasmocitoides.
Rasgos clínicos: Edad superior a 40 años, usualmente debuta con afectación de la médula ósea, ganglios linfáticos y/o bazo. Característico componente M en suero (cuadro clínico de macroglobulinemia de Waldenstrom), y síntomas derivados de hiperviscosidad. Ocasional crioglobulinemia. Es posible la demostración de infección por Virus de la Hepatitis C en un porcentaje de casos. Se trata de una enfermedad indolente, no curable, que con frecuencia progresa a transformación a linfoma de célula grande.
Morfología: Patrón difuso, en ocasiones interfolicular. Citología compuesta por linfocitos pequeños, linfocitos plasmocitoides (con amplio citoplasma similar al de las células plasmáticas), células plasmáticas e inmunoblastos. Se suelen identificar Cuerpos de Dutcher (inclusiones nucleares) (fig. 5).
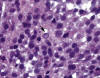
Fig. 5: Inmunocitoma con presencia de
cuerpos de Dutcher nucleares (flecha).
Es importante la ausencia de los rasgos diagnósticos de otros procesos linfoproliferativos incluyendo LLC-B, LCM, LF, LZM-MALT o LEZM. La presencia de seudofolículos es característica de LLC-B, y no debe hacerse el diagnóstico de inmunocitoma en estos casos. Los linfomas en mucosas o piel con rasgos de inmunocitoma forman parte del espectro de diferenciación que puede verse en linfomas de la zona marginal.
Inmunofenotipo y genotipo: IgM, IgG, IgA de supercicie y citoplasmática. CD23 -/+, CD5 -, CD43+/-.
Una alteración genética característica de alguno de estos procesos es la translocación que afecta al gen PAX5, localizado en 9p13. Estos tumores tienen un moderado grado de mutación somática del gen de las inmunoglobulinas.
Variante: Enfermedad de cadenas pesadas gamma.
LINFOMA FOLICULAR (LF)
Tumor derivado de linfocitos del centro germinal, caracterizado por la sobreexpresión de la proteína Bcl2 secundaria a la translocación t(14; 18).
Rasgos clínicos: La edad media de presentación es superior a 30 años, aunque pueden verse ocasionales formas juveniles. Frecuentemente se trata de una enfermedad diseminada al diagnóstico, con afectación ganglionar y de médula ósea y/o bazo. Es un tumor indolente, no curable. La probabilidad de supervivencia depende del estadio clínico y grado histológico. Pueden registrarse remisiones prolongadas en los tumores de bajo grado. Hasta un 60% de los casos llegan a mostrar transformación a linfoma de células grandes, que suele estar asociada a inactivación de los genes p53 y p16 u otras alteraciones citogenéticas.
Morfología: Prácticamente todos lo casos muestran de forma más o menos patente un patrón folicular, aunque esto puede afectar a tan sólo una fracción del ganglio biopsiado. Ocasionalmente, la demostración del patrón folicular puede requerir inmunotinción para células dendríticas (CD21 o CD23). La citología muestra siempre una mezcla de centrocitos y centroblastos, si bien en proporción variable. Además hay abundantes células dendríticas foliculares y linfocitos T entremezclados con el tumor, hasta el punto que la abundancia de células T puede llegar a enmascarar el cuadro. Otro hallazgo frecuente es la presencia de abundante esclerosis, especialmente en localización retroperiotoneal.
Los LFs se gradúan histológicamente de acuerdo a la proporción relativa de centroblastos:
Grado I: predominio de centrocitos.
Grado II: con proporción de centroblastos (<15 por campo de gran aumento).
Grado III: predominio de centroblastos (>15 por campo de gran aumento).
Los diferentes estudios de correlación clínica muestran que el grado III se correlaciona con una conducta clínica más agresiva, si bien su reconocimiento tiene una baja reproducibilidad. Específicamente se ha encontrado que el tiempo libre de fallo es mas corto en estos linfomas foliculares grado III, aunque la supervivencia global no parecer diferir significativamente. Desde un punto de vista práctico se agrupan los casos de grado I y II como tumores de bajo grado histológico, mientras que los casos de grado III corresponden a tumores de alto grado.
Inmunofenotipo y genotipo: CD10+, Bcl6+, CD23 -, CD43 -, CD5 -, IgD -. El 85% de los casos muestra alta expresión de Bcl2, en contraste con la imagen usual de los centros germinales reactivos. Esta expresión de Bcl2 se ha convertido clásicamente en un útil rasgo diagnóstico, que permite identificar fases precoces de linfoma folicular. De hecho, la expresión simultánea de Bcl2 y Bcl6 (fig. 6) puede considerarse un rasgo neoplásico característico, ya que no se observa en ningún estadio madurativo B en condiciones fisiológicas.
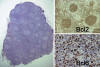
Fig. 6: Linfoma folicular. Patrón nodular
y coexpresión de Bcl2 y Bcl6 por las células tumorales.
La inmunotinción para marcadores de células dendríticas foliculares revela la presencia de una malla densa soportando las células tumorales. Otros rasgos característicamente descritos en los folículos neoplásicos son su bajo índice proliferativo mediante Ki67 y la ausencia de células del manto mediante inmunotinción para IgD.
La mayoría de los casos (alrededor del 90%) de LF tiene una translocación t(14; 18) y un pequeño porcentaje tiene amplificación del gen BCL2. Sin embargo, la ausencia de translocación de BCL2 y la ausencia de expresión de proteína mediante inmunohistoquímica son hallazgos comunes en linfomas foliculares de localización cutánea.
Es característico de estos tumores la presencia de abundantes mutaciones somáticas y mutaciones en curso del gen IgH (así como de otros genes como Bcl6 o C-MYC), del mismo modo que sucede con las células centrofoliculares en condiciones normales.
Variantes:
Los casos con un patrón difuso son excepcionales, y probablemente representan otro tipo tumoral, a menos que muestren un fenotipo y genotipo característico, con expresión de Bcl6, CD10 y Bcl2.
LF cutáneo. Desde un punto de vista de presentación clínica y evolución se trata de un tumor similar a linfomas cutáneos de la zona marginal, si bien la morfología y fenotipo de los mismos muestra que estos tumores derivan de células del centro germinal. No se detecta t(14;18) en las formas primarias restringidas a piel y es importante excluir la presencia de la translocación, ya que suele estar asociada con enfermedad diseminada.
Variantes morfológicas: La presencia de células monocitoides no es rara en estos tumores, y no justifica por sí misma un diagnóstico de linfoma B de la zona marginal. También puede verse diferenciación plasmocitoide y células en anillo de sello.
LINFOMA DE CÉLULAS DEL MANTO (LCM)
Tumor derivado de linfocitos vírgenes del manto folicular, caracterizados por sobreexpresión de Ciclina D1 como consecuencia de la translocación t(11; 14).
Rasgos clínicos: En la mayoría de los casos, se trata de una enfermedad diseminada al diagnóstico o brevemente tras el mismo. Edad superior a 40 años. Aunque la mayoría de los casos debuta con afectación ganglionar, la afectación inicial puede ser extraganglionar (poliposis linfomatosa intestinal, amígadalas) o esplénica. Es una enfermedad moderadamente agresiva, no curable.
Morfología: Patrón vagamente nodular, o tipo zona del manto (rodeando centros germinales reactivos preexistentes). Citología monomorfa, constituida por linfocitos pequeños de núcleo hendido, semejantes a centrocitos, aunque variaciones en la fijación del tejido pueden alterar de forma significativa la morfología. Frecuentemente existen histiocitos epitelioides salpicados que pueden dar una imagen de «cielo estrellado».
Inmunofenotipo y genotipo: CD23 -, CD10-, CD43 +, CD5 +, IgD +, Bcl6-, CD10-. También es típica la demostración de una malla laxa de células dendríticas, que recuerda la existente en el manto folicular no neoplásico. La demostración Inmunohistoquímica de Ciclina D1 nuclear presente en la mayoría de las células neoplásicas es muy característica de este tipo de linfoma (fig. 7).
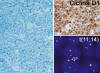
Fig. 7: Patrón difuso en un linfoma de
células del manto. La sobreexpresión nuclear de Ciclina D1 es secundaria a la
translocación t(11;14) (FISH en sección de parafina).
Estudios moleculares han mostrado que la sobreexpresión de Ciclina D1 es secundaria a t(11; 14), presente en el 100% de estos casos. Sin embargo, las técnicas basadas en PCR para la detección de esta alteración tienen una sensibilidad raramente superior al 40%, por lo que es recomendable el uso de técnicas de citogenética convencional o FISH (fig. 7), con sensibilidad próxima al 100%.
Por otro lado, a pesar de que la ausencia de mutaciones somáticas del gen IgH es un rasgo característico de las células del manto en condiciones fisiológicas, en los LCM se detectan mutaciones somáticas en aproximadamente 1/3 de los casos y al contrario de lo que ocurre en la LLC-B, la presencia de mutaciones somáticas de IgH no parece guardar correlación con el pronóstico. Mutaciones o deleciones del gen ATM también se están describiendo en un alto porcentaje de estos casos. A pesar de tratarse de una enfermedad en general agresiva, la combinación de diferentes alteraciones moleculares puede determinar las diferencias que se observan en la evolución clínica.
Variantes:
Existe una variante agresiva, denominada blastoide, con citología blástica o anaplásica o pleomórfica, con frecuente aneuploidía, asociada con mutaciones de p53 e inactivación de p16, dotada de un curso clínico más agresivo. La demostración de la sobreexpresión de Ciclina D1 es crítica para este diagnóstico.
Existe también una variante indolente (10-15% de los casos), en alguno de los casos asociada a un patrón tipo «zona del manto»
Variantes morfológicas: La presencia de cordones de células claras (a veces llamadas monocitoides), en ganglio linfático o bazo, no invalida el diagnóstico de linfoma del manto. Aunque clásicamente se ha postulado la citología monomorfa de los LCM, estos tumores pueden mostrar un cierto grado de variabilidad citológica, más acentuada cuando las células tumorales infiltran centros germinales, donde se encuentran células neoplásicas con mayor tamaño y ocasional citología tipo centroblasto.
LINFOMA DE LA ZONA MARGINAL DEL TEJIDO LINFOIDE ASOCIADO A
MUCOSAS (MALT)Linfoma extraganglionar derivado de linfocitos de la zona marginal que han sufrido alteraciones moleculares convergentes en la ruta de señalización celular NF-kappaB, bloqueando la ruta conducente a apoptosis.
Rasgos clínicos: Es una enfermedad de adultos, con ligero predominio de mujeres. Con relativa frecuencia, existe un antecedente de enfermedad autoinmune o inflamatoria como síndrome de Sjogren, tiroiditis de Hashimoto o gastritis asociada a Helicobacter pylori. La mayoría de los casos se presenta como enfermedad localizada, aunque ha sido también descrita afectación extraganglionar múltiple.
Morfología: Tumores de origen extraganglionar que incluyen células B de la zona marginal, células monocitoides, linfocitos pequeños, y frecuentemente un componente de células plasmáticas (habitualmente monoclonales) y blastos salpicados. El tumor parece originarse en la localización de la zona marginal, para extenderse después hacia el folículo linfoide, donde crea imágenes de colonización folicular que pueden simular al linfoma folicular. Las células neoplásicas al infiltrar estructuras glandulares, suelen formar las características lesiones linfoepiteliales.
Estos tumores con frecuencia muestran transformación a linfoma de célula grande. Si el tumor tiene áreas difusas de linfoma de célula grande, debe ser etiquetado como tal, haciendo notar su asociación con linfoma LZM-MALT. Hay datos indicando que estos tumores de mayor agresividad responden peor a terapias dirigidas a erradicación de Helicobacter pylori.
Inmunofenotipo y genotipo: Las células neoplásicas muestran un fenotipo CD20+ CD5- CD23- CD43-/+, CD10-, bcl6-, ciclinaD1-. Como en otras linfomas de baja fracción de crecimiento, el tumor es Bcl2+.
Se detecta con relativa frecuencia trisomía 3, aunque el hallazgo más característico es la presencia de t(11; 18) (q21;q21), presente en alrededor del 30% de los casos, y que podría distinguir un subgrupo de pacientes con un comportamiento más agresivo y menor probabilidad de respuesta al tratamiento erradicador de H. pylori en tumores de localización gástrica. Más recientemente han sido también descritas las translocaciones t(1;14)(p22;q32) afectando al gen Bcl10, o t(14;18)(q32;q21) afectando a MALT1. Todas estas alteraciones citogenéticas convergen en producir activación de la ruta NF-kappaB, bloqueando así la apoptosis. La presencia de mutación somática de los genes IgH es también un hallazgo característico de estas neoplasias.
LINFOMA GANGLIONAR DE LA ZONA MARGINAL (LZM)
Tumor ganglionar con similar morfología y fenotipo que los linfomas de la zona marginal tipo MALT extranodales.
Rasgos clínicos: Edad>40 años. En una enfermedad ganglionar, frecuentemente contigua con una localización mucosa (típicamente adenopatía cervical). Se describe frecuentemente asociación con síndrome de Sjögren.
Morfología: Patrón nodular, con colonización folicular. Es importante en el reconocimiento de este tumor la presencia de manguitos de células B monocitoides periféricos a los folículos linfoides, aunque este no es un hallazgo exclusivo de esta neoplasia, pudiendo verse también en LF y otros. La citología tumoral está formada por linfocitos de tamaño medio con citoplasma claro, algunos inmunoblastos salpicados y ocasional diferenciación plasmacítica.
Inmunofenotipo y genotipo: CD23 -, CD10-, CD43 -, CD5 -, IgD -, Ciclina D1 -. Ausencia de t(11; 14), o t(14; 18). Relativa frecuencia de trisomía 3. La ausencia de marcadores moleculares o fenotípicos característicos dificulta el diagnóstico preciso, que descansa muy importantemente en los rasgos arquitecturales del tumor. Podría tratarse de une entidad heterogénea, y de hecho numerosos estudios moleculares parecen confirmar esta presunción.
LINFOMA ESPLÉNICO DE LA ZONA MARGINAL (LEZM)
Linfoma de linfocitos pequeños, con sintomatología derivada de la afectación esplénica. Las células tumorales pueden identificarse en sangre periférica (fig. 8) como linfocitos vellosos, justificando la sinonimia empleada ocasionalmente en la literatura (FAB: linfoma esplénico con linfocitos vellosos).

Fig. 8: Linfoma esplénico de la zona
marginal. Linfocitos vellosos en sangre periférica. Infiltración esplénica con
patrón micronodular. Infiltración de médula ósea con la característica invasión
intrasinusoidal (CD20).
Rasgos clínicos: Edad>50 años. Esplenomegalia, con grado variable de linfocitosis y pancitopenia. Enfermedad indolente, no curable, con buena respuesta a la esplenectomía. Un porcentaje (15-20%) de los casos sufre transformación agresiva, ligada a mutaciones de p53.
Morfología: Rasgos característicos de la afectación esplénica son el patrón micronodular, con reemplazamiento de los folículos linfoides preexistentes, con una citología polimorfa que incluye linfocitos pequeños, blastos y células de la zona marginal. Aunque la presencia de cordones de células marginales es típica de la afectación esplénica, estos no suelen verse en el ganglio linfático. La médula ósea suele mostrar afectación intertrabecular, con típica presencia de cordones neoplásicos en la luz de los sinusoides (fig. 8). Puede observarse en ganglios periféricos progresión a linfoma de células grandes en una fracción de
Inmunofenotipo y genotipo: CD23 -, CD10-, CD43 -, CD5 -, IgD +, Ciclina D1 -. Patrón en diana de tinción con Ki67 (identificando una zona proliferante alrededor de centros germinales reactivos preexistentes).
Ausencia de t(11; 14), o t(14; 18). Se describen alteraciones genéticas múltiples en los cromosomas 1,3 y 7. Frecuentes pérdidas en la región 7q31-7q32, que diferencia esta enfermedad de otros linfoproliferativos. Presencia de mutaciones somáticas de los genes IgVH en 50% de los casos, caracterizados por una conducta más indolente.
Variantes: Un pequeño porcentaje de casos puede mostrar ausencia de linfocitos vellosos en sangre periférica. Pueden observarse variantes con patrón difuso y recientemente se ha reconocido también una variante agresiva con incremento en el porcentaje de blastos, expresión de Ciclina D1 y/o mutaciones de p53.
LINFOMA B DIFUSO DE CÉLULAS GRANDES (LBDCG)
Mas que de una entidad precisa se trata de un grupo de linfomas B agresivos, en el que no existen conocimientos precisos que permitan una subclasificación de este frecuente grupo de linfomas. Incluye localizaciones ganglionares y extraganglionares, enfermedades primarias y secundarias.
Rasgos clínicos: Edad>50 años, aunque pueden verse en niños y adultos jóvenes. Con relativa frecuencia el tumor tiene un estadio clínico limitado, especialmente en los de presentación extraganglionar. Enfermedad agresiva, curable después de tratamiento, con una esperanza de vida a los 5 años cercana al 50% de los pacientes.
Morfología: El patrón arquitectural de estos tumores es difuso en la mayoría de los casos, aunque la presencia de algunos nódulos es también posible, siempre que la mayoría del tumor muestre un patrón difuso. La afectación ganglionar puede ser parcial, sinusoidal o interfolicular.
La citología puede estar compuesta por células tipo centroblasto, inmunoblasto o células grandes pleomórficas (fig. 9). El diagnóstico diferencial con linfomas T siempre requiere estudio inmunofenotípico y molecular.
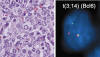
Fig. 9: Linfoma B difuso de células
grandes con citología de tipo centroblástico y reordenamiento de Bcl6 (FISH en
sección de parafina).
En casos ocasionales pueden mostrar un alto índice proliferativo y patrón en cielo estrellado requiriendo adicionalmente estudio citogenético o molecular para excluir la posibilidad de linfoma de Burkitt. Algunos de estos casos han sido denominados en el pasado como linfomas Burkitt-like, denominación que en la actualidad no es reconocida como categoría diagnóstica por la OMS.
Inmunofenotipo y genotipo: Fenotipo B con ocasional ausencia de algún antígeno Pan-B. Expresión de Bcl6 en hasta el 80% de estos casos. La expresión de Bcl2 es más frecuente en los tumores de origen ganglionar. Esta expresión elevada de Bcl2 se asocia además a una menor supervivencia global y especialmente a supervivencia libre de enfermedad más corta. La alta expresión de p53 se ha encontrado asociada, como en otros linfomas, a una pobre respuesta a quimioterapia y supervivencias más cortas. Un alto índice proliferativo aparece, en numerosas series, también asociado a una supervivencia global más corta. No se ha encontrado que la expresión de CD30 guarde relación con ninguna otra característica del tumor.
Puede detectarse translocación de Bcl2 con IgH en un porcentaje aproximado del 20% de casos. Un porcentaje mayor (alrededor del 30%) tiene translocaciones afectando la región 3q27 (Bcl6) (figura 9), con mayor frecuencia en casos con origen extraganglionar. Además de translocaciones afectando a Bcl6, otras alteraciones moleculares pueden participar también en la expresión alterada de este proto-oncogen.
Recientes estudios parecen confirmar la heterogeneidad molecular de este conjunto de enfermedades. Así, mediante análisis de expresión génica se han descrito las categorías de LBDCG de tipo «centro germinal» y de tipo «célula B activada», que parecen diferir en su patogenia y respuesta terapéutica.
Variantes: Los LBDCG de localización extraganglionar, aunque no sean diferenciables morfológicamente de sus contrapartidas nodales, deben de identificarse como tales ya que numerosos estudios han confirmado que estos tumores presentan unas características clinicopatológicas dependientes de la localización de la enfermedad. Por ejemplo, la conducta clínica de los tumores que se presenta en la piel es extremadamente indolente, lo cual contrasta con tumores de citología similar que se pueden presentar en el sistema nervioso central.
Linfomas cutáneos de células grandes B son tumores con muy baja agresividad clínica, en los que aún existe muy escasa documentación acerca de sus características moleculares o fenotípicas. Algunos datos preliminares que muestran que la expresión de Bcl2 es característica de los linfomas B cutáneos de células grande originados en las piernas, con comportamiento más agresivo, esperan su confirmación por parte de otros grupos.
Linfoma B de células grandes gastrointestinal: En estos tumores (como en los cutáneos) con frecuencia se observa una transición con un componente de célula pequeña, sugiriendo que algunos de estos tumores derivan de linfomas de bajo grado preexistentes (tipo MALT). También el hecho de que los linfomas de célula grande gastrointestinales compartan la frecuencia de trisomía 3 e inestabilidad de microsatélites propias de linfomas MALT, sugiere un origen compartido para estas neoplasias. Adicionalmente, el origen en tejido linfoide asociado a mucosas puede explicar parcialmente la conducta clínica menos agresiva de muchos de estos casos.
Si embargo, algunas de las localizaciones que se asocian con formas características de esta enfermedad son reconocidas como entidades diagnósticas por la OMS:
Mediastínico: Típicamente es un tumor de adultos jóvenes (más frecuente en mujeres que hombres), con sintomatología clínica derivada de su localización (ocasionalmente síndrome de vena cava superior). Con frecuencia estos tumores tienen una esclerosis fina que abraza y deforma las células neoplásicas, dificultando su reconocimiento. De forma peculiar, se describe la frecuente expresión de CD30, CD23 y proteína MAL1 en este tipo tumoral. La presencia de ganancias del cromosoma 9 y amplificación del oncogén REL soporta la existencia de rasgos característicos de esta neoplasia. El curso clínico es estrechamente dependiente del estadio clínico.
Intravascular: El linfoma B intravascular tiene una distribución histológica fundamentalmente limitada la luz de pequeños vasos, sobre todo capilares. Típicamente el tumor afecta vasos del SNC o piel, aunque puede verse en cualquier otra localización extraganglionar. Consecuentemente el cuadro clínico que produce es bastante singular, y muy dependiente del órgano afecto. La enfermedad suele tener un curso clínico extremadamente agresivo, agudo, que conduce rápidamente a la muerte. Se observan ocasionalmente casos de similar morfología pero fenotipo T.
Linfoma primario asociado a efusiones: Se trata de linfomas más frecuentes en enfermos HIV+, aunque pueden verse también casos en pacientes sin immunodepresión. Se presentan como derrames en cavidades corporales (con más frecuencia derrame pleural o ascítico). La citología es característica, de tipo plasmablástico. La mayoría de los casos están asociados al virus KSHV y EBV.
Granulomatosis linfomatoide: Se trata de un proceso linfoproliferativo angiocéntrico y angiodestructivo, afectando de forma característica al pulmón con una clínica y radiología peculiares, que en la actualidad se reconoce como una forma de linfoma B de células grandes inducido por EBV (fig. 10). Habitualmente tiene una heterogénea composición celular «rico en células T». También se puede presentar en otras localizaciones extranodales como riñón, piel, cerebro o tracto gastrointestinal.
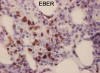
Fig. 10: Granulomatosis
linfomatoide. Infiltrado celular polimorfo con presencia de EBV en las células B
neoplásicas.
Variantes citológicas: Se pueden identificar en función de la composición celular predominante: centroblástico, inmunoblástico, linfoma B rico en células T e histiocitos, y formas anaplásicas. Algunas variedades morfológicas presentan correlación definida con características clínicas o moleculares de estos tumores. Así la forma de linfoma B rico en células T suele mostrar una presentación clínica diseminada, con frecuente afectación hepatoesplénica. Tumores de citología inmunoblástica tienen un comportamiento más agresivo, mientras que linfomas en los que el tipo citológico está compuesto por células B multilobuladas con frecuencia expresan altos niveles de Bcl2 y derivan de células centrofoliculares. Se han descrito linfomas B de localización intrasinusoidal, con citología tipo inmunoblasto, con expresión de la proteína ALK citoplasmática. No obstante, los intentos de crear una clasificación basada en variedades morfológicas tropiezan con la dificultad de la existencia de formas intermedias y la baja reproducibilidad de la misma.
LINFOMA DE BURKITT (LB)
Tumor derivado de células del centro germinal con muy alta fracción de crecimiento secundaria a sobreexpresión de C-MYC derivada de translocaciones afectando a 8q24. EBV puede encontrarse en un porcentaje variable de casos.
Rasgos clínicos: Enfermedad más frecuente en niños y adultos jóvenes, con mayor frecuencia en varones. La forma no endémica suele manifestarse como enfermedad abdominal, con afectación intestinal, mesentérica y en ocasiones genitourinaria. No obstante, la enfermedad puede debutar en ganglio linfático, siendo esta presentación más común en adultos. Los linfomas de Burkitt asociados a inmunodeficiencia (HIV y otros) se manifiestan con mayor frecuencia como enfermedad ganglionar. La mayoría de las leucemias agudas de las denominadas en el pasado como morfología L3 corresponden a linfoma de Burkitt.
Morfología: Tumor de frecuente localización extraganglionar, con citología cohesiva de tamaño medio, contorno nuclear relativamente redondeado, varios nucleolos paramediales, citoplasma basofílico con vacuolas, alto índice mitótico (fig. 11).
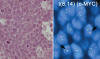
Fig. 11: Linfoma de Burkitt. Determinación
de reordenamiento de c-MYC mediante FISH (secciones de parafina).
En ocasiones el origen del tumor se encuentra en centros germinales periféricos al tumor principal. Existen frecuentes variaciones morfológicas de este patrón (citología grande, pleomorfismo, diferenciación plasmocitoide), que hacen frecuentemente necesario el concurso de técnicas de citogenética o moleculares para el diagnostico.
Inmunofenotipo y genotipo: La mayoría de los casos de linfoma de Burkitt tienen un fenotipo CD20+ CD10+ bcl6+ bcl2- Tdt-. La negatividad para bcl2 y Tdt es un criterio exigible en este diagnóstico. En la mayoría de los casos es posible encontrar translocaciones del gen c-myc con IgH, lambda o kappa. La detección de la translocación es actualmente relativamente simple empleando técnicas basadas en FISH, y es imprescindible en todos los casos de diagnóstico dudoso por la trascendencia pronostica y terapéutica del diagnóstico diferencial con otros linfomas agresivos. El punto de ruptura en el gen IgH en los casos endémicos afecta la región J, mientras que en los casos esporádicos la translocación afecta la región switch. La presencia de EBV caracteriza los casos endémicos y un porcentaje del 25 al 40% de los asociados con inmunodeficiencia. La presencia de mutaciones somáticas del gen IgH confirma que estos tumores tienen un origen postfolicular.
Variantes: Algunos de los linfomas denominados en el pasado como Burkitt-like, especialmente en niños e inmunodeficientes, deben de ser identificados como linfomas de Burkitt, ya que la conducta clínica, fenotipo y genotipo se superpone por completo con esta enfermedad. La negatividad para bcl2 y una fracción de crecimiento del 100% son criterios necesarios para el diagnóstico de linfoma de Burkitt.
BIBLIOGRAFÍA
Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994; 84: 1361-92.
Melnyk A, Rodriguez A, Pugh WC, Cabannillas F. Evaluation of the Revised European-American Lymphoma classification confirms the clinical relevance of immunophenotype in 560 cases of aggressive non-Hodgkin's lymphoma. Blood. 1997; 89: 4514-20.
A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood. 1997; 89: 3909-18.
Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol. 1998; 16: 2780-95.
Jaffe ES HN, Stein H and Vardiman JW. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. World health organization classification of tumours. Lyon: IARC Press.; 2001.
Quintanilla-Martinez L, Zukerberg LR, Ferry JA, Harris NL. Extramedullary tumors of lymphoid or myeloid blasts. The role of immunohistology in diagnosis and classification. Am J Clin Pathol. 1995; 104: 431-43.
Potter MN, Cross NC, van Dongen JJ, et al. Molecular evidence of minimal residual disease after treatment for leukaemia and lymphoma: an updated meeting report and review. Leukemia. 1993; 7: 1302-14.
Sole F, Woessner S, Perez-Losada A, et al. Cytogenetic studies in seventy-six cases of B-chronic lymphoproliferative disorders. Cancer Genet Cytogenet. 1997; 93: 160-6.
Juliusson G, Oscier DG, Fitchett M, et al. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med. 1990; 323: 720-4.
Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003; 348: 1764-75.
Ritgen M, Lange A, Stilgenbauer S, et al. Unmutated immunoglobulin variable heavy-chain gene status remains an adverse prognostic factor after autologous stem cell transplantation for chronic lymphocytic leukemia. Blood. 2003; 101: 2049-53.
Brito-Babapulle V, Ellis J, Matutes E, et al. Translocation t(11;14)(q13;q32) in chronic lymphoid disorders. Genes Chromosomes Cancer. 1992; 5: 158-65.
Schlette E, Bueso-Ramos C, Giles F, Glassman A, Hayes K, Medeiros LJ. Mature B-cell leukemias with more than 55% prolymphocytes. A heterogeneous group that includes an unusual variant of mantle cell lymphoma. Am J Clin Pathol. 2001; 115: 571-81.
Miranda RN, Briggs RC, Kinney MC, Veno PA, Hammer RD, Cousar JB. Immunohistochemical detection of cyclin D1 using optimized conditions is highly specific for mantle cell lymphoma and hairy cell leukemia. Mod Pathol. 2000; 13: 1308-14.
Bosch F, Campo E, Jares P, et al. Increased expression of the PRAD-1/CCND1 gene in hairy cell leukaemia. Br J Haematol. 1995; 91: 1025-30.
Fonseca R, Hoyer JD, Aguayo P, et al. Clinical significance of the translocation (11;14)(q13;q32) in multiple myeloma. Leuk Lymphoma. 1999; 35: 599-605.
Perez-Simon JA, Garcia-Sanz R, Tabernero MD, et al. Prognostic value of numerical chromosome aberrations in multiple myeloma: A FISH analysis of 15 different chromosomes. Blood. 1998; 91: 3366-71.
Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003; 30: 110-5.
Iida S, Rao PH, Ueda R, Chaganti RS, Dalla-Favera R. Chromosomal rearrangement of the PAX-5 locus in lymphoplasmacytic lymphoma with t(9;14)(p13;q32). Leuk Lymphoma. 1999; 34: 25-33.
Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003; 101: 1220-35.
Martinez-Climent JA, Alizadeh AA, Segraves R, et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood. 2003; 101: 3109-17.
Hans CP, Weisenburger DD, Vose JM, et al. A significant diffuse component predicts for inferior survival in grade 3 follicular lymphoma, but cytologic subtypes do not predict survival. Blood. 2003; 101: 2363-7.
Franco R, Fernandez-Vazquez A, Mollejo M, et al. Cutaneous presentation of follicular lymphomas. Mod Pathol. 2001; 14: 913-9.
Peng HZ, Du MQ, Koulis A, et al. Nonimmunoglobulin gene hypermutation in germinal center B cells. Blood. 1999; 93: 2167-72.
Camacho FI, Algara P, Rodriguez A, et al. Molecular heterogeneity in MCL defined by the use of specific VH genes and the frequency of somatic mutations. Blood. 2003; 101: 4042-6.
Martinez N, Camacho FI, Algara P, et al. The molecular signature of mantle cell lymphoma reveals multiple signals favoring cell survival. Cancer Res. 2003; 63: 8226-32.
Ott G, Katzenberger T, Greiner A, et al. The t(11;18)(q21;q21) chromosome translocation is a frequent and specific aberration in low-grade but not high-grade malignant non-Hodgkin's lymphomas of the mucosa-associated lymphoid tissue (MALT-) type. Cancer Res. 1997; 57: 3944-8.
Rosenwald A, Ott G, Stilgenbauer S, et al. Exclusive detection of the t(11;18)(q21;q21) in extranodal marginal zone B cell lymphomas (MZBL) of MALT type in contrast to other MZBL and extranodal large B cell lymphomas. Am J Pathol. 1999; 155: 1817-21.
Ye H, Liu H, Attygalle A, et al. Variable frequencies of t(11;18)(q21;q21) in MALT lymphomas of different sites: significant association with CagA strains of H pylori in gastric MALT lymphoma. Blood. 2003; 102: 1012-8.
Ye H, Dogan A, Karran L, et al. BCL10 expression in normal and neoplastic lymphoid tissue. Nuclear localization in MALT lymphoma. Am J Pathol. 2000; 157: 1147-54.
Lucas PC, Yonezumi M, Inohara N, et al. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. 2001; 276: 19012-9.
Campo E, Miquel R, Krenacs L, Sorbara L, Raffeld M, Jaffe ES. Primary nodal marginal zone lymphomas of splenic and MALT type. Am J Surg Pathol. 1999; 23: 59-68.
Nathwani BN, Anderson JR, Armitage JO, et al. Marginal zone B-cell lymphoma: A clinical comparison of nodal and mucosa-associated lymphoid tissue types. Non-Hodgkin's Lymphoma Classification Project. J Clin Oncol. 1999; 17: 2486-92.
Berger F, Felman P, Thieblemont C, et al. Non-MALT marginal zone B-cell lymphomas: a description of clinical presentation and outcome in 124 patients. Blood. 2000; 95: 1950-6.
Arcaini L, Paulli M, Boveri E, Magrini U, Lazzarino M. Marginal zone-related neoplasms of splenic and nodal origin. Haematologica. 2003; 88: 80-93.
Camacho FI, Algara P, Mollejo M, et al. Nodal marginal zone lymphoma: a heterogeneous tumor: a comprehensive analysis of a series of 27 cases. Am J Surg Pathol. 2003; 27: 762-71.
Conconi A, Bertoni F, Pedrinis E, et al. Nodal marginal zone B-cell lymphomas may arise from different subsets of marginal zone B lymphocytes. Blood. 2001; 98: 781-6.
Camacho FI, Garcia JF, Sanchez-Verde L, et al. Unique phenotypic profile of monocytoid B cells: differences in comparison with the phenotypic profile observed in marginal zone B cells and so-called monocytoid B cell lymphoma. Am J Pathol. 2001; 158: 1363-9.
Catovsky D, Matutes E. Splenic lymphoma with circulating villous lymphocytes/splenic marginal-zone lymphoma. Semin Hematol. 1999; 36: 148-54.
Camacho FI, Mollejo M, Mateo MS, et al. Progression to large B-cell lymphoma in splenic marginal zone lymphoma: a description of a series of 12 cases. Am J Surg Pathol. 2001; 25: 1268-76.
Mollejo M, Menarguez J, Lloret E, et al. Splenic marginal zone lymphoma: a distinctive type of low-grade B-cell lymphoma. A clinicopathological study of 13 cases. Am J Surg Pathol. 1995; 19: 1146-57.
Isaacson PG, Matutes E, Burke M, Catovsky D. The histopathology of splenic lymphoma with villous lymphocytes. Blood. 1994; 84: 3828-34.
Franco V, Florena AM, Campesi G. Intrasinusoidal bone marrow infiltration: a possible hallmark of splenic lymphoma. Histopathology. 1996; 29: 571-5.
Audouin J, Le Tourneau A, Molina T, et al. Patterns of bone marrow involvement in 58 patients presenting primary splenic marginal zone lymphoma with or without circulating villous lymphocytes. Br J Haematol. 2003; 122: 404-12.
Sole F, Woessner S, Florensa L, et al. Frequent involvement of chromosomes 1, 3, 7 and 8 in splenic marginal zone B-cell lymphoma. Br J Haematol. 1997; 98: 446-9.
Sole F, Salido M, Espinet B, et al. Splenic marginal zone B-cell lymphomas: two cytogenetic subtypes, one with gain of 3q and the other with loss of 7q. Haematologica. 2001; 86: 71-7.
Hernandez JM, Garcia JL, Gutierrez NC, et al. Novel genomic imbalances in B-cell splenic marginal zone lymphomas revealed by comparative genomic hybridization and cytogenetics. Am J Pathol. 2001; 158: 1843-50.
Mateo M, Mollejo M, Villuendas R, et al. 7q31-32 allelic loss is a frequent finding in splenic marginal zone lymphoma. Am J Pathol. 1999; 154: 1583-9.
Martinez-Climent JA, Sanchez-Izquierdo D, Sarsotti E, et al. Genomic abnormalities acquired in the blastic transformation of splenic marginal zone B-cell lymphoma. Leuk Lymphoma. 2003; 44: 459-64.
Algara P, Mateo MS, Sanchez-Beato M, et al. Analysis of the IgV(H) somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood. 2002; 99: 1299-304.
Dunn-Walters DK, Boursier L, Spencer J, Isaacson PG. Analysis of immunoglobulin genes in splenic marginal zone lymphoma suggests ongoing mutation. Hum Pathol. 1998; 29: 585-93.
Mateo MS, Mollejo M, Villuendas R, et al. Molecular heterogeneity of splenic marginal zone lymphomas: analysis of mutations in the 5' non-coding region of the bcl-6 gene. Leukemia. 2001; 15: 628-34.
Mollejo M, Algara P, Mateo MS, et al. Splenic small B-cell lymphoma with predominant red pulp involvement: a diffuse variant of splenic marginal zone lymphoma? Histopathology. 2002; 40: 22-30.
Lloret E, Mollejo M, Mateo MS, et al. Splenic marginal zone lymphoma with increased number of blasts: an aggressive variant? Hum Pathol. 1999; 30: 1153-60.
Gruszka-Westwood AM, Hamoudi RA, Matutes E, Tuset E, Catovsky D. p53 abnormalities in splenic lymphoma with villous lymphocytes. Blood. 2001; 97: 3552-8.
Lossos IS, Akasaka T, Martinez-Climent JA, Siebert R, Levy R. The BCL6 gene in B-cell lymphomas with 3q27 translocations is expressed mainly from the rearranged allele irrespective of the partner gene. Leukemia. 2003; 17: 1390-7.
Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003; 101: 2914-23.
Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002; 346: 1937-47.
Chan JK. Mediastinal large B-cell lymphoma: new evidence in support of its distinctive identity. Adv Anat Pathol. 2000; 7: 201-9.
Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood. 1996; 88: 645-56.
Haralambieva E, Banham AH, Bastard C, et al. Detection by the fluorescence in situ hybridization technique of MYC translocations in paraffin-embedded lymphoma biopsy samples. Br J Haematol. 2003; 121: 49-56.
Dalla-Favera R, Lombardi L, Pelicci PG, Lanfrancone L, Cesarman E, Neri A. Mechanism of activation and biological role of the c-myc oncogene in B-cell lymphomagenesis. Ann N Y Acad Sci. 1987; 511: 207-18.
Hecht JL, Aster JC. Molecular biology of Burkitt's lymphoma. J Clin Oncol. 2000; 18: 3707-21.
![]()