
Vol. 37, n.º 4, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 4, 2004 |
María Martino González, Javier Gómez Román, Francisco Mazorra Macho, Fernando Val-Bernal
Dpto. de Anatomía Patológica. Hospital Universitario Marqués de Valdecilla. Santander. marmartino@hotmail.com
RESUMEN
Presentamos el caso de una mujer con un síndrome de Sjögren diagnosticada, por estudio de sangre periférica y de la médula ósea, de «Linfoma de zona marginal, compatible con linfoma esplénico de linfocitos vellosos» a la que se practicó esplenectomía.
El estudio posterior anatomopatológico del bazo confirmó la presencia de un linfoma esplénico de zona marginal de bajo grado, pero además existía otro linfoma difuso de células grandes, de fenotipo B, de alto grado de malignidad, sincrónico con el anterior y que los estudios previos hematológicos no habían detectado. Esta asociación es muy poco frecuente.
Este caso demuestra la necesidad de la integración de los datos clínicos, patológicos y moleculares para llegar al diagnóstico correcto.
Palabras clave: Linfoma de la zona marginal, linfoma de alto grado, bazo, sincrónico.
SUMMARY
We report a case of a marginal zone lymphoma consistent with an splenic lymphoma of hairy lymphocytes diagnosed in peripheral blood and bone marrow in a 69-years old female with a Sjögren syndrome. Splenectomy was indicated..
A later microscopic examination of spleen confirmed the presence of a low grade splenic lymphoma. However, another large cell diffuse lymphoma of phenotype B of high grade of malignancy, synchronic with the former was found. Previous hematologic studies had not detected it.
This case shows the necessity of integrate clinical, pathologic and molecular biology information to get a correct diagnosis.
Key words: Marginal zone lymphoma, high-grade lymphoma, spleen, synchronous.
INTRODUCCIÓN
Los linfomas esplénicos de la zona marginal son neoplasias linfoides de bajo grado de malignidad, de fenotipo B generalmente de supervivencia larga incluso cuando infiltran la médula ósea. Suponen menos del 1% de los tumores linfoides. Sin embargo su frecuencia puede ser mayor ya que, al ser un tumor con expresión en sangre periférica —linfocitos vellosos circulantes—, algunos casos de leucemia linfoide crónica inclasificables, CD5 negativos, podrían corresponder en realidad a linfomas esplénicos de la zona marginal. Afectan por igual a pacientes de ambos sexos por encima de la quinta década de la vida (1).
Como no suelen responder bien a la quimioterapia, e incluso algunos son refractarios a ella, y sí a la esplenectomía, éste es el tratamiento de elección, describiéndose supervivencias largas.
Clínicamente existe una asociación bien documentada con enfermedades autoinmunes del tipo de Síndrome de Sjögren (2).
Como en otros casos de neoplasias linfoides, en el curso de su evolución, se ha descrito la transformación en linfomas de alto grado de malignidad y también, aunque es más raro, desarrollo de forma sincrónica de un linfoma esplénico de la zona marginal y un linfoma de células grandes de alto grado de malignidad (1,2).
Presentamos una paciente con un linfoma de la zona marginal y un linfoma de alto grado de malignidad de células grandes, que aparecieron de forma sincrónica en el bazo de una mujer de 69 años de edad y discutimos el valor de la biopsia quirúrgica para el diagnóstico de las neoplasias de estirpe linfoide.
HISTORIA CLÍNICA
Mujer de 69 años cuyos antecedentes personales más relevantes eran: hipercolesterolemia, hipertensión arterial, osteoartrosis y osteoporosis. Hace años se sometió a una histerectomía y doble anexectomía por quistes ováricos.
Hace ocho años comenzó con sequedad ocular además de la artrosis referida, por lo que se realizaron pruebas de laboratorio encaminadas a descartar enfermedad autoinmune, entre las que destacaba un título de anticuerpos antinucleares de 1/320, que si bien no era muy elevado, indicaba sospecha de Síndrome de Sjögren primario que se confirmó finalmente mediante una biopsia de glándula salival (Se observaron tres focos de más de 50 linfocitos/4 mm2, con fibrosis focal). Otros anticuerpos determinados en esa época (antiDNA nativo, antiRNA, complemento: C3 y C4, antiDNA de cadena única (SS), inmunoglobulinas: A, G, M, y E y crioglobulinas) fueron negativos.
Enfermedad actual: Desde hace tres semanas, presenta un cuadro de febrícula (37,5 °C), astenia, anorexia, disminución de peso, tos no productiva, dolor pleurítico en costado izquierdo, dolor en hipogastrio y sudoración profusa nocturna.
Exploración física: Pálida. Abdomen: Blando, doloroso en epigastrio e hipogastrio sobre todo en fosa iliaca izquierda. Resto normal.
Pruebas complementarias:
Hemograma: 11.500 leucocitos micro/l (4,8-10,8 leucocitos micro/l) , linfocitos 19,5 % (20,0- 51,0%), hematíes 3,43 x 106 micro/l (4,50-6,20 x 106 micro/l), hemoglobina 9,1 g/dl (13,5-18,0 g/dl), hematocrito 28,3% (42,0-52,0%) y actividad de protrombina: 63% (70-100%). Velocidad de sedimentación en 1 hora 110 mm. El resto era normal.
Bioquímica general: LDH 709 U/L (230-460 U/L), GOT 58 U/L (1-31 U/L), GPT 69 U/L (1-31 U/L), GAMMA-GT 103 U/L (7-32 U/L) , fosfatasa alcalina 172 U/L (40-129 U/L) y proteína C reactiva 15,5 mg/dl (0,1-0,5 mg/dl), siendo el resto de los parámetros normales.
Marcadores tumorales: CA 125: 37,2 (1,0-17,2) y Beta 2 microglobulina: 3,54 (1,20-2,50). Resto normal.
Proteinograma: Alfa 2 globulinas ligeramente elevadas: 1,2 gr/dl (0,5-0,8).
Ecografía abdominal: Lesión esplénica de 10 cm heterogénea con vasos en su interior.
Tomografía axial computerizada abdominal: Tres lesiones esplénicas ocupantes de espacio, heterogéneas con necrosis en su interior.
Mielograma: Sangre periférica: El 48% población linfoide (5% población total) son linfocitos B con expresión parcial de CD11c, CD5, CD23 y CD43. Expresan con intensidad CD20 y FMC7. Son negativos frente a CD30, CD10, CD38, CD56 y CD103. Se observa expresión de cadena ligera kappa, con restricción de cadena ligera lambda.
Médula ósea: El 5,6% de la población total corresponde a población clonal B con el fenotipo similar al estudiado en sangre periférica.
El diagnóstico fue de «Linfoma de zona marginal» con intensa expresión de CD20 (compatible con linfoma esplénico de linfocito velloso), ante lo que se procede a realizar una esplenectomía.
MATERIAL Y MÉTODOS
Se recibió, en el Departamento de Anatomía Patológica, un bazo de 720 gramos (Normal: hasta 200 gramos) que medía 14 x 14 x 6,5 cm. Externamente, se encontraba deformado periféricamente por dos nódulos blanquecinos. Al corte, los nódulos medían 7,2 y 3,8 cm respectivamente de diámetro máximo. Eran sólidos, con bordes irregulares, de consistencia elástica, y de coloración blanquecina (fig. 1). Se tomaron muestras para congelación tanto de la lesión como del parénquima «respetado» y el resto se fijó en formol tamponado neutro al 10% durante 24 horas.
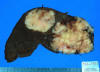
Fig. 1: Corte macroscópico del bazo donde
se observan dos nódulos tumorales en el seno de un parénquima aparentemente
normal.
Tras su inclusión en parafina, se realizaron cortes que se tiñeron con técnicas de rutina: Hematoxilina-Eosina, PAS, Gordon-S, así como las siguientes técnicas inmunohistoquímicas (tabla 1) para lo que se utilizó un inmunoteñidor automático «Techmate 500» (Biotek; Santa Barbara, CA) con el método de «Envision +», revelándose con diaminobenzidina y hematoxilina de Mayer.

Para los estudios de Patología Molecular, se seleccionó tejido del parénquima esplénico y de los nódulos tumorales para la extracción de ADN, haciéndose el análisis de reordenamiento clonal del gen de las cadenas pesadas de las inmunoglobulinas (región FR2-JH) y el del gen TCR gamma (región VJ), mediante cuantificación genómica (PCR) región específica y análisis heteroduplex. Como control de la integridad del ADN se utilizó una región altamente conservada del exón 5 del gen p53 y en todas las reacciones se incluyeron controles negativos para asegurar la ausencia de contaminación.
RESULTADOS
En los cortes de las lesiones nodulares, microscópicamente se observó una proliferación difusa de células grandes, con elevado índice mitótico, con núcleos vesiculosos nucleolos prominentes y citoplasma bien definido (fig. 2). Inmunohistoquímicamente resultaron positivas para CD45, CD20, bcl-6 y CD30 (fig. 3), con alto índice proliferativo con MIB-1 (fig. 4). Esta lesión estaba bien delimitada. El resto del parénquima esplénico mostraba una proliferación de células linfoides pequeñas de núcleos arriñonados y escaso citoplasma (fig. 5), predominantemente afectando a la pulpa blanca —en la zona marginal—, y focalmente infiltrando la pulpa roja, que expresaban fenotípicamente CD45, CD20, CD43, IgD kappa y bcl-2 (fig. 6), con un índice proliferativo bajo. Había además un componente de células plasmocitoides y plasmáticas mezcladas con las citadas.
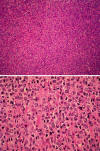
Fig. 2: Proliferación monomorfa y difusa
de células linfoides grandes (HE 4X). En la imagen inferior, detalle a gran
aumento de estas células con núcleos vesiculosos, nucleolos prominentes y
citoplasma bien definido (HE 40X).
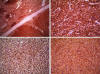
Fig. 3: Panel inmunohistoquímico donde se
demuestra el fenotipo del linfoma difuso de células grandes. La expresión del
bcl-6 y CD 30, se aproxima al 100% de las células tumorales (CD45: 4X. CD20:
10X. Bcl-6: 10X y CD30: 10X).
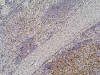
Fig. 4: En el ángulo inferior derecho, el
linfoma de células grandes con un índice proliferativo alto que contrasta con la
baja actividad proliferativa del linfoma de células pequeñas, en la parte
superior de la fotografía. MIB-1 (Ki 67) (10X).

Fig. 5: Proliferación predominantemente
nodular del linfoma de células pequeñas, que se expande desde la zona marginal
hacia la pulpa roja esplénica (HE 4X).
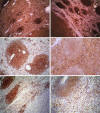
Fig. 6: Perfil inmunohistoquímico del
linfoma de células pequeñas. Los distintos anticuerpos resaltan su patrón
predominantemente nodular. Obsérvese la ausencia de expresión de bcl-2 en el
linfoma de alto grado, en el ángulo inferior derecho (CD45 y CD20: 4X. IgD,
kappa, bcl-2 y CD43: 10X).
En los cortes de la zona de transición entre los dos linfomas, se observa una separación neta entre los mismos, claramente visible con el Ki-67(MIB-1), lo que indicaba el distinto comportamiento biológico de ambos en lo que a actividad proliferativa se refiere (fig. 4).
Existía reordenamiento clonal del gen de la IgH (región FR2-JH), tanto en la población de células grandes como en la de linfocitos «monocitoides» de la zona marginal que confirmaba la existencia de dos poblaciones neoplásicas de estirpe B, es decir con el mismo fenotipo, pero distintas en lo que se refiere a la morfología y actividad proliferativa. El reordenamiento clonal del gen TCR gamma (segmento VJ) fue negativo en los dos bloques (fig. 7).
![]()
Fig. 7: Patología Molecular: En la calle
1: Marcador de peso molecular. Calle 2: Linfoma de alto grado sin análisis de
heterodúplex . Calle 3: Linfoma de alto grado con análisis de heterodúplex.
Calle 4: Control interno del linfoma de alto grado exón 5 de p53. Calle 6:
Linfoma de bajo grado sin análisis de heterodúplex. Calle 7: Linfoma de bajo
grado con análisis de heterodúplex. Calle 8 : Control interno del linfoma de
bajo grado exón 5 de p53.
El diagnóstico fue de Linfoma difuso de célula grande de fenotipo B (CD45, CD20 y bcl-6) con alto índice proliferativo, expresión de CD30 y p53 negativo sincrónico con un linfoma de célula pequeña de fenotipo B de la zona marginal esplénica (CD45, CD20, CD43, IgD kappa y bcl-2), con bajo índice proliferativo.
DISCUSIÓN
La zona marginal es hoy reconocida como uno de los compartimentos B dependientes de los tejidos linfoides, situada siempre externamente a la región del manto folicular . Está muy bien desarrollada en órganos sometidos a alta circulación de antígenos como son la pulpa blanca del bazo, placas de Peyer intestinales, amígdalas, etc. y existen algunos procesos reactivos, como la toxoplasmosis donde existe una expansión obvia de esta zona microanatómica ganglionar. Al ser una encrucijada de caminos en la que confluyen células que viajan hacia los centros germinales y también desde éstos hacia los senos, las células que se observan a este nivel suelen ser de varios tipos como linfocitos pequeños, células grandes similares a inmunoblastos B, macrófagos, granulocítos y células plasmáticas que están mezcladas con las células que verdaderamente pertenecen a este compartimento, las denominadas células B de la zona marginal, caracterizadas por citoplasma pálido relativamente abundante, y núcleo central que recuerda al de los centrocitos del centro germinal por lo que también se las ha denominado «linfocitos centrocitoides» y también «linfocitos B monocitoides» por sus núcleos en ocasiones arriñonados y su localización subcapsular sinusoidal como los macrófagos/monocitos (2,3).
Inmunohistoquímicamente expresan marcadores de fenotipo B (CD20, CD19, CD79a), salvo CD5, CD10 y CD23 y además expresan bcl-2 e IgM (más que IgA e IgG) con expresión variable de IgD (positiva en los linfocitos esplénicos de la zona marginal y negativa en los linfocitos de la zona marginal en tejido linfoide asociado a mucosas (MALT) y linfocitos de la zona marginal nodal). Otros antígenos expresados por estas células son CD21 y CD35, C3 y fosfatasa alcalina. Tampoco expresan ciclina-D1. La expresión de CD43 es variable (1,2,3).
Estudios en roedores han demostrado que los linfocitos de la zona marginal juegan un papel timo-independiente tipo 2 (TI-2), en la respuesta contra antígenos como polisacáridos capsulares bacterianos con una reacción muy rápida y efectiva precisamente derivada de su localización estratégica, actuando estas células en vanguardia de la respuesta inmune (2). Este hecho se refleja también en la intensidad variable de actividad según el territorio y así son más activas las zonas marginales de las placas de Peyer y de los ganglios, que las del bazo, reflejando una mayor ó menor exposición antigénica local. Esta variabilidad se ha demostrado también a nivel genético, no solo en los linfocitos no tumorales de este compartimento, sino parece que la refleja también su contrapartida neoplásica, el linfoma de la zona marginal, que muestra lugares mutados, hipermutados e incluso casos sin mutaciones, en la región variable de las cadenas pesadas de las inmunoglobulinas (VH), en algunos casos publicados dónde se ha estudiado dicha región (2,4)
Estos fenómenos tan dinámicos que ocurren en este compartimento ha llevado a estudiar sus células en relación con señales que controlan la adhesión y motilidad celulares. En este sentido se ha estudiado una tirosin-kinasa, Pyk-2, que actúa integrando estas señales utilizando ratones knockout Pyk-2 negativos que carecen específicamente de células de la zona marginal esplénicas con déficit de respuestas IgG3 a antígenos tipo TI-2. Se produce en ellos de entrada, un defecto en la motilidad linfocitaria y baja respuesta a una citoquina segregada por los macrófagos residentes (células mieloides-monocitoides que cohabitan con las células de la zona marginal) en este territorio, que sería un miembro de la familia TNF, denominado BlyS (estimulador de linfocitos B) que, en condiciones normales, induciría una rápida diferenciación de células de la zona marginal hacia células secretoras de inmunoglobulinas (Igs.). Pues bien, cuando en estos ratones se inyecta BlyS aumentan el número de células de la zona marginal y también los niveles de Igs. y también se activa el factor de transcripción NF-(kappa)B, esencial para el desarrollo, supervivencia y proliferación de las células de la zona marginal (5-7). Además la sobreexpresión de BlyS se acompaña de la aparición de anticuerpos patógenos autoinmunes tipo lupus. Se puede pensar que si BlyS se disregula y/ó se mantiene en el tiempo, de ello puede derivarse la diferenciación hacia estados patogénicos, bien de tipo autoinmune (lupus) bien de tipo tumoral como los linfomas (2,5). En nuestro caso existía una enfermedad autoinmune previa (síndrome de Sjögren). También es bien conocida la mayor frecuencia que tienen los pacientes con enfermedades autoinmunes para desarrollar linfomas, sobre todo de la zona marginal.
Existe cierta controversia en la clasificación de los linfomas de la zona marginal. La clasificación REAL los dividió en Linfomas de la zona marginal «Extranodales» ó de tipo MALT (asociados a mucosas) y Linfomas de la zona marginal «Nodales» (que representan los «linfomas monocitoides»), contemplando una entidad provisional, el Linfoma de la zona marginal «Esplénico», pendiente de clasificar (8). Esta tercera entidad se separó definitivamente en la clasificación de la OMS: el Linfoma de la zona marginal Esplénico (denominado por algunos autores como linfoma esplénico con linfocitos vellosos) (1).
Estas tres entidades están separadas en virtud de sus diferentes comportamientos clinico-patológicos, considerándose enfermedades diferentes, como contempla la OMS en su clasificación (1,2).
El linfoma esplénico de zona marginal, considerado un linfoma extranodal, es un tumor de bajo grado y curso indolente, con una media de supervivencia del 78% a los 5 años (3). Microscópicamente crece de forma nodular desde la pulpa blanca expandiendo al principio la zona marginal periférica y luego invade la pulpa roja adyacente, aunque reciéntemente se ha publicado una posible variante difusa de este linfoma (9). Es conocida sin embargo la posible progresión a linfomas de alto grado, la mayoría entre los 12 y 85 meses después del diagnóstico del linfoma de bajo grado y se ha relacionado esta circunstancia con mutaciones en p53 y con índices de proliferación elevados. La aparición de forma sincrónica de los dos linfomas, uno de la zona marginal de bajo grado y el otro de células grandes de alto grado, es rara: En una de las series más extensas publicadas sólo 2 de 12 casos eran sincrónicos y de ellos sólo uno mostró una presentación clínica limitada al bazo, semejante a nuestro caso (3).
En nuestra paciente, el linfoma de alto grado mostraba un alto índice proliferativo (Ki67) sin sobreexpresión de p53 y sin embargo expresaba CD30, marcador de mal pronóstico relacionado con los linfomas anaplásicos. La kinasa del linfoma anaplásico (ALK), resultó negativa. Si bien el término «Linfoma Anaplásico» se reserva en la actualidad a un tumor con características clinico-patológicas bien definidas, de fenotipo T, CD30 y ALK, también es cierto que existen linfomas de fenotipo B de alto grado que expresan CD30 y son de mal pronóstico.
Este caso como otros ilustra una vez más que, aunque es posible el diagnóstico de algunos linfomas y procesos linfoproliferativos (leucemias, mielomas) con métodos citológicos hematológicos, sigue siendo esencial el estudio anatomopatológico de estos procesos, sobre todo en órganos sólidos dónde una mera punción, aunque da información muy fiable de la morfología celular aislada, la aleja absolutamente de su ambiente arquitectural que, es sabido que es esencial para diagnosticar de forma cierta muchos tipos de linfomas porque, el material utilizado en punciones muchas veces no representa ni cuantitativa ni cualitativamente todo el proceso patológico ni aún en el caso de aplicación a este material, de técnicas sofisticadas de inmunocitoquímica y/ ó biología molecular (10-12).
En la literatura están apareciendo muchos falsos negativos y falsos positivos en muestras pequeñas de ganglios linfáticos y de otros tejidos (11,12), de forma aislada y/ó sin tener en cuenta el lugar anatómico donde están situadas estas células ni su morfología (centros germinales de ganglios linfáticos con hiperplasias foliculares reordenan de forma clonal muchas veces para las cadenas de las inmunoglobulinas) cuando se aplican técnicas de biología molecular. Existen centros germinales con mayor diámetro que el de una aguja de las utilizadas en PAAF (punción-aspiración) y la escasez de material puede hacer que se diagnostique erróneamente un linfoma. Por otro lado, como es nuestro caso, la muestra puede reflejar sólo una de las lesiones, el linfoma de bajo grado de malignidad, no habiendo detectado el linfoma más agresivo, de alto grado de malignidad, adyacente (11).
La patología linfoide es un paradigma de cómo debe enfocarse el estudio con grupos multidisciplinarios que permitan avanzar en el estudio y conocimiento de este grupo de tumores. Siguen siendo esenciales la integración de datos clínicos, morfológicos celulares y topográficos, inmunohistoquímicos y de patología molecular, para su diagnóstico, como hemos demostrado en este caso.
BIBLIOGRAFÍA
Jaffe ES, Harris NL, Stein H et al. Tumors of Haematopoietic and Lymphoid Tissues. Pathology & Genetics (135-137). World Health Organization (WHO). Ed. IARC Press. Lyon 2001.
Maes B and De Wolf-Peeters C. Marginal zone cell lymphoma, an update on recent advances. Histopathology 2002; 40: 117-26.
Camacho F, Mollejo M, Mateo M et al. Progression to large B cell lymphoma in splenic marginal zone lymphoma: A description of a series of 12 cases. Am J Surg Pathol. 2001; 25: 1268-76.
Zhu D, Orchard J, Oscier DG et al. Vh gene analysis of splenic marginal zone lymphomas reveals diversity in mutational status and initiation of somatic mutation in vivo. Blood 2002; 100: 2659-61.
Guinamard R, Okigaki M, Schlessinger J et al. Absence of marginal zone B cells in Pyk-2-deficent mice defines their role in the humoral response. Nature Immunol. 2000; 1: 31-6.
Cariappa A, Liou H, Horwitz H et al. Nuclear factor kB is required for the development of marginal zone B lymphocytes. J. Exp. Med. 2000; 192: 1175-82.
Gross J, Johnston J, Mudri S et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature 2000; 404: 995-9.
Harris NL, Jaffe ES, Stein H et al. A revised european-american classification of lymphoid neoplasms: a proposal from the international lymphoma study group. Blood 1994; 84: 1361-92.
Mollejo M, Algara P, Mateo M et al. Splenic small B-cell lymphoma with predominant red pulp involvement: a diffuse variant of splenic marginal zone lymphoma?. Histopathology 2002; 40: 22-30.
Wright PA, Wynford-Thomas D. The polymerase chain reaction: miracle or mirage? A critical review of uses and limitations in diagnosis and research. J. Pathology 1990; 162: 99-117.
Zhou XG, Sandvej K, Gregersen N et al. Detection of clonal B cells in microdissected reactive lymphoproliferations: A possible diagnostic pitfalls in PCR analysis of immunoglobulin heavy chain gene rearrangement. Molecular Pathol 1999; 52: 104-10.
Nihal M, Mikkola D, Wood GS. Detection of clonally restricted immunoglobulin heavy chain gene rearrangements in normal and lesional skin: Analysis of the B cell component of the skin-associated lymphoid tissue and implications for molecular diagnosis of cutaneous B cell lymphomas. J Molecular Diagnostics 2000; 2: 5-10.
![]()