
Vol. 38, n.º 1, 2005
|
REVISTA
ESPAÑOLA DE
Vol. 38, n.º 1, 2005 |
ORIGINALES
María Virginia Ortega1, Gabriela Gualco1, María Luisa Musto, Richard Wins2, José Maggiolo2, Gonzalo Ardao1
Hospital Central de las Fuerzas Armadas.
Montevideo. Uruguay.
1 Servicio de Anatomía Patológica.
2 Servicio de Cirugía de Tórax.
virortega@adinet.com.uy
RESUMEN
El tumor fibroso solitario de topografía pleural (TFSP) es una neoplasia infrecuente, probablemente originada en el tejido conectivo submesotelial. Dichas neoplasias crecen dentro del espacio pleural, usualmente unidos a la hoja visceral de la pleura por un pedículo. Clínica e imagenológicamente pueden ser confundidos con mesoteliomas localizados o tumores primitivos de pulmón. Histológicamente están constituidos por células fusiformes, con grados variables de celularidad y de tejido extracelular. El perfil inmunohistoquímico característico, con positividad para vimentina y CD34 junto a la negatividad para citoqueratina, permite distinguirlo del mesotelioma maligno. Los factores considerados de buen pronóstico son: presencia de pedículo, circunscripción o encapsulamiento, tamaño menor a 10 cm, resección completa y ausencia de invasión pulmonar. Los criterios histológicos de malignidad incluyen: hipercelularidad, más de 4 mitosis por 10 CGA, pleomorfismo celular, necrosis y hemorragia. Se ha reportado que los tumores con sobreexpresión de la proteína p53 y con un elevado porcentaje de células neoplásicas Ki 67 positivas, se asocian a un pobre pronóstico. Presentamos 3 casos de TFSP, 2 de ellos con características clínico-patológicas de comportamiento benigno, que fueron hallazgos incidentales en exámenes radiológicos. El caso restante se manifestó con un cuadro clínico de osteoartropatía pulmonar hipertrofiante, mostrando en la histología características de malignidad.
Palabras claves: tumor fibroso solitario, pleura, inmunohistoquímica.
SUMMARY
Solitary fibrous tumours of the pleura (SFTP) are uncommon neoplasms. Such well circumscribed tumors arise usually from submesothelial tissue and may have a pedicle attached to the pleura. Generally asymptomatic, they usually have a benign behavior. Clinical and radiological findings suggest peripheral lung tumours or pleural mesothelioma. Histopathologically they are characterized by the presence of spindle-shaped or oval cells with variable cellularity, scattered haphazardly among a collagenized stroma. Usually positive for CD34 and vimentin, they are negative for cytokeratin. Histological indicators of malignancy are high cellularity, more than 4 mitotic figures per 10 HPF, nuclear pleomorphism and hemorrhage. Good prognosis is associated with presence of a pedicle, circumscription, size less than 10 cm, complete resection and absence of lung invasion. Overexpression of p53 and high label index of Ki 67 are associated with poor outcome. We analyzed three SFTP, two with benign clinical and morphological characteristics and one with histopathological indicators of malignancy and pulmonary osteoarthropathy at diagnosis.
Key words: solitary fibrous tumor, pleural tumor, immunohistochemical assays.
INTRODUCCIÓN
La mayoría de los tumores fibrosos solitarios de topografía pleural (TFSP) tienen un comportamiento biológico benigno, aunque raramente pueden recurrir localmente o dar metástasis (1-3). Las recurrencias pueden ser tardías, con un período de latencia de hasta 30 años (2). Las formas malignas pueden ser de novo o resultar de la transformación de un TFS benigno (4). Su presentación radiológica y la heterogeneidad morfológica que presentan, plantean diagnósticos diferenciales con lesiones pleuropulmonares malignas y benignas. La consulta intraoperatoria generalmente no arriba a un diagnóstico definitivo, ya que es necesario un muestreo amplio de la lesión y la realización de técnicas de inmunohistoquímica (IHQ) para la confirmación diagnóstica. El tratamiento de elección de estas neoplasias es quirúrgico, con resección completa de toda la lesión (1). Se presentan tres casos de TFSP y se analizan las características clínico-patológicas diagnósticas y de carácter pronóstico.
MATERIAL Y MÉTODOS
Los casos clínicos (fig. 1) se resumen en la tabla 1. Las piezas de resección fueron fijadas en formalina tamponada al 10%. Se tomó para histología 1 fragmento por cada cm de tumor, muestreando toda la lesión y su vinculación con el parénquima adyacente. Se procesaron de manera rutinaria con inclusión en parafina. Se colorearon con hematoxilina - eosina (HE), Alcian-Blue-PAS, y con técnica para fibras de reticulina. Se realizó marcación inmunohistoquímica (IHQ) con los siguientes anticuerpos: citoqueratina cocktail (AE1/AE3) (DAKO), CD34 (IMMUNOTECH), vimentina (DAKO), desmina (DAKO), proteína S-100 (IMMUNOTECH), actina de músculo liso (HHF35) (DAKO), CD31 (DAKO) y p53 (IMMUNOTECH), utilizando el método estreptavidina biotina peroxidasa y revelando con diaminobencidina como cromógeno.
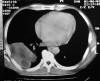
Fig. 1: Caso
3. Tomografía axial computarizada (TAC): gran proceso sólido pulmonar de lóbulo
inferior izquierdo con amplio contacto parietal.
RESULTADOS
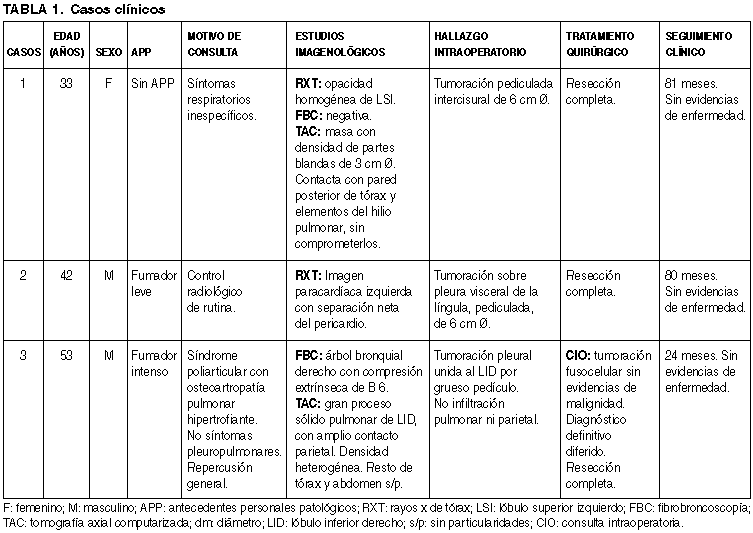
Macroscopía. Los tres casos presentaron aspecto similar, correspondiendo a tumoraciones circunscriptas, con presencia de pedículo en los casos 1 y 2. Dimensiones: el caso 1 midió 6 ´ 6 ´ 2,5 cm; el segundo caso 6 ´ 4 ´ 3 cm y el tercer caso 15 ´ 10 ´ 8 cm. La superficie externa era lisa, brillante, con aspecto seroso. Superficie de corte sólida, firme, blanquecina con focos parduscos, y en sectores aspecto arremolinado (fig. 2).

Fig. 2: Caso
2. Tumoración sólida, circunscripta, blanco-grisácea, sin áreas de necrosis ni
hemorragia, adherida a pulmón por grueso pedículo.
Microscopía. Todos los casos se caracterizaron por una proliferación fusocelular, con patrón arquitectural vorticelar, en «esqueleto de arenque», y sectores con haces entrelazados sin un patrón definido. El caso 2 presentó áreas con patrón Kaposi-símil. La densidad celular era variable en cada tumor, alternando con sectores de esclerosis difusa. En los casos 1 y 2 no se observó pleomorfismo celular ni atipías nucleares. No se observaron mitosis en el caso 1. El caso 2 presentó 3 mitosis por 10 campos de gran aumento (CGA). En el caso 3 se apreciaban sectores densamente celulares, con atipías nucleares, presencia de células gigantes multinucleadas y elevado índice mitótico (9 mitosis por 10 CGA), así como también extensas áreas de necrosis isquémica.
Inmunohistoquímica (IHQ). Los hallazgos con técnicas de inmunohistoquímica se resumen en la tabla 2 (figura 3).


Fig. 3: Caso
2. TFSP con inmunomarcación citoplasmática intensa y difusa para CD34 (DAB 10x).
DISCUSIÓN
Los Tumores fibrosos solitarios son neoplasia muy poco frecuentes, con una incidencia reportada de 2,8 casos por 100.000 tumores (1). Representan el 5% de los tumores pleurales (1,3) y constituyen el segundo tumor primitivo más frecuente en dicha topografía, después del mesotelioma maligno difuso (1,4). En menor frecuencia se pueden observar en otras topografías (mediastino, pulmón, tracto respiratorio superior, hígado, peritoneo, tiroides, mama, próstata, etc.). Pueden aparecer a cualquier edad, incluyendo la niñez con una media de 51 años, sin diferencias entre ambos sexos (5). A diferencia del mesotelioma, no se asocian con la exposición al asbesto u otro carcinógeno (6,7). Los TFSP generalmente constituyen un hallazgo incidental en estudios radiológicos, presentándose como nódulos pulmonares periféricos bien circunscriptos, de densidad radiológica homogénea, asintomáticos en 50% de los casos (8). Los síntomas más frecuentes son tos, disnea y dolor torácico. Las lesiones grandes pueden dar lugar a hipoglicemia o derrame pleural; raramente galactorrea (5,6,8,9). La hipoglicemia y la osteoartropatía pulmonar hipertrófica, esta última presente en el 10% de los pacientes, que constituyó la presentación clínica del caso 3, se asocian con un fenotipo maligno (1,10). Los tres casos reportados presentaron aspecto macroscópico típico, pero se describen casos con crecimiento endofítico hacia el parénquima pulmonar subyacente, o más raramente con crecimiento infiltrante, dando el aspecto de lesiones parenquimatosas (3,5,6,8-11). Histológicamente consisten, en forma característica, en una proliferación sólida fusocelular, que presenta áreas alternantes de hiper e hipocelularidad, con patrón arquitectural estoriforme y áreas de esclerosis difusa (2,5,6,8-12). Sin embargo pueden presentar sectores con otros patrones arquitecturales, simulando otras neoplasias mesenquimáticas, como ser: áreas hemangiopericitoma símil, fibrosarcoma símil, sarcoma de Kaposi símil, sectores fusocelulares sin patrón definido y áreas de aspecto epitelioide (que presentan el perfil IHQ de las células fusocelulares) (2,5,8-10,12,13). El perfil inmunofenotípico característico del TFS es la positividad uniforme e invariable para vimentina; positividad también uniforme y en más del 80% de los casos para CD34; y negatividad para la expresión de citoqueratina (CK), como se observó en los 3 casos analizados. Recientemente se ha descrito que expresan CD99 y bcl 2. Raramente pueden expresar desmina y actina de músculo liso. Otros marcadores como actina muscular específica, actina de músculo esquelético, enolasa neuronal específica, antígeno de membrana epitelial, CD31, factor VIII, proteína S-100 y calretinina, son negativos (5,8,9,12-17). Se puede observar marcada disminución o pérdida completa de la expresión de CD34 en los focos de transformación maligna (1,4). No hay unanimidad de criterios con respecto a las características predictoras de una conducta agresiva del tumor. England et al (2) establecieron que la hipercelularidad, el pleomorfismo nuclear y la presencia de más de 4 figuras mitóticas por 10 campos de gran aumento, son indicadores histológicos de comportamiento maligno. Sin embargo la mayoría de los parámetros histológicos mencionados no siempre se han correlacionado en forma directa con la sobrevida (2,5,6,11), ya que algunos tumores pediculados e histológicamente malignos se han considerado curados con la resección completa (2,4-6). En algunas series se ha reportado que el tamaño mayor a 10 cm y una localización atípica del tumor (pleura parietal, mediastino, o crecimiento endofítico dentro del pulmón) se asocian a recurrencias locales y curso clínico fatal (10,18,19). Por lo tanto, los factores asociados a buen pronóstico son: la presencia de pedículo, la circunscripción o encapsulación, la resección completa, el tamaño menor a 10 cm, junto a la ausencia de invasión pulmonar, actividad mitótica y pleomorfismo celular (2,5,6,10). Más recientemente se evaluó la expresión de Ki 67 y p53 mediante IHQ (1,4), con el fin de identificar otros factores predictivos de comportamiento. El antígeno de proliferación Ki 67 mostró un bajo índice de proliferación en todos los casos histológicos benignos, independientemente del tamaño tumoral (4). Por el contrario, las lesiones histológicamente malignas mostraron alto índice de proliferación (mayor al 20%) (4,10). La sobreexpresión de p53 se observó en todos los casos malignos que presentaron un curso clínico fatal o recurrencias locales, así como atipía nuclear, elevada actividad mitótica o invasión local (4,10). Los casos de TFS benignos estudiados fueron negativos (4). Estos hallazgos, sin embargo, deben ser confirmados con series mayores (1,19). El principal diagnóstico diferencial es el mesotelioma maligno, principalmente sus variantes sarcomatoide y desmoplásica, sobre todo en mesoteliomas localizados, en TFSP sin pedículo, relacionado al pulmón o la pared costal, y principalmente en material biópsico (8,12). En estos casos la IHQ es muy importante, ya que los mesoteliomas son CK y generalmente calretinina positivos. Los otros diagnósticos diferenciales que se plantean son: timoma fusocelular, fibrosarcoma, fibromatosis, sarcoma sinovial, histiocitoma fibroso maligno, neurofibroma, neurofibrosarcoma bien diferenciado, schwannoma, tumores de músculo liso, angiosarcoma, sarcoma de Kaposi, hemangiopericitoma y carcinoma fusocelular metastásico. Para descartar estas neoplasias es necesario realizar un panel de IHQ que incluya varios marcadores, ya que algunos de ellos pueden expresar en forma variable positividad para CD34 (5,8,9,10,14-16,18,19,20).
En estas neoplasias, por lo tanto, es necesario realizar un análisis preciso macro y microscópico, que incluya técnicas de inmunohistoquímicas, para la confirmación diagnóstica e identificación de criterios de malignidad.
Los tres casos analizados correspondieron a TFSP con características morfológicas y perfil IHQ típicos. Los casos 1 y 2 presentaron todos los factores de buen pronóstico mencionados. El caso restante presentó características histológicas e IHQ de malignidad, junto a la presentación clínica de osteoartropatía pulmonar hipertrófica. Los tres casos continúan vivos y asintomáticos, con un seguimiento de 81 y 80 meses en los casos 1 y 2, respectivamente, y 24 meses en el caso 3. Dado que es posible que existan recurrencias tardías, es necesario un seguimiento a largo plazo de estos pacientes, aún en las lesiones que muestran características de buen pronóstico.
BIBLIOGRAFÍA
Brozzetti S, D´Andrea N, Limiti MR, Pisanelli MC, De Angelis R, Cavallaro. A. Clinical Behavior of Solitary Tumors of the Pleura. An Immunohistochemical Study. Anticancer Res 2000; 20: 4701-6.
England DM, Hochholzer L, and McCarthy MJ. Localized Benign and Malignant Fibrous Tumors of the Pleura. A clinicophatologic review of 223 cases. Am J Surg Pathol 1989; 13: 640-58.
Nonaka M, Kadokura M, Takaba T. Benign Solitary Fibrous Tumor of the Parietal Pleura which Invaded the Intercostal Muscle. Lung Cancer 2001; 31: 325-9.
Yokoi T, Suzuki T, Yatabe Y, Suzuki M, Kurumayas H, Koshikawa T, et al. Solitary fibrous tumour: significance of p53 and CD 34 immunoreactivity in its malignant transformation. Histopathol 1998; 32: 423-32.
Moran CA, Suster S, Koss MN. The Spectrum of Histologic Growth Patterns in Benign and Malignant Fibrous Tumors of the Pleura. Semin Diagn Pathol 1992; 9: 169-80.
Briselli M, Marck EJ, Dickersin GR. Solitary Fibrous Tumors of the Pleura: eight new cases and review of 360 cases in the literature. Cancer 1981; 47: 2678-89.
Morgan MB, Smoller BR. Solitary Fibrous Tumors are immunophenotypically distinct from mesotheliomas. J Cutan Pathol 2000; 27:451-4.
Van Rijm M, Lombard CM, Rouse RV. Expression of the CD34 by Solitary Fibrous Tumors of the Pleura, Mediastinum, and Lung. Am J Surg Pathol 1994; 18: 814-20.
Mc Guire LLJ, Chan HS. Pang J. Case Report. Solitary Fibrous Tumor Of the Pleura: Expression of Citokeratins. Pathol 1990; 22: 232-4.
Rodriguez H, Vicuña R, Rodríguez A, Gómez A, Chávez L, Lazos M, et al. Tumor fibroso solitario. Estudio de 15 casos, 13 de localización atípica y dos con hipoglicemia. Patología 2002; 40: 87-97.
Caruso RA, La Spada F, Gaeta M, Minutoli I, Inferrera C. Report of an Intrapulmonary Solitary Fibrous Tumor: Fine-Needle Aspiration Cytologic Findings, Clinicopathological and Immunohistochemical Features. Diagn Cytopathol 1996; 14: 64-70.
Flint A, Weiss SW. CD-34 and Keratin Expression Distinguishes Solitary Fibrous Tumors (Fibrous Mesothelioma) of Pleura from Desmoplastic Mesothelioma. Hum Pathol 1995; 26: 428-31.
Hanau CA, Miettinen M. Solitary Fibrous Tumor: Histological and Immunohistochemical Spectrum of Benign and Malignant Variants Presenting at Diferent Sites. Hum Pathol 1995; 26: 440-9.
Marchevsky AM, Varshney D, Fuller C. Mediastinal epithelioid solitary fibrous tumor. Arch Pathol Lab Med 2003; 127: e212-5.
Proca D, Ross P, Pratt J, Frankel WL. Smooth muscle tumor of the pleura. A case report and review of the literature. Arch Pathol Lab Med 2000; 124: 1688-92.
Shidam VB, Chivukula M, Gupta D, Rao RN, Komorovski R. Immunohistochemical comparison of gastrointestinal stromal tumor and solitary fibrous tumor. Arch Pathol Lab Med 2002. 126: 1189-92.
Van Rijn M and Rouse RV. CD34 a review. Appl Immunohistochem 1994; 2: 71-80.
Tihan T, Viglione M, Rosemblum MK, Olivi A, Burger P. Solitary fibrous tumors in the central nervous system. A clinicopathologic review of 18 cases and comparison to meningeal hemangiperycitomas. Arch Pathol Lab Med 2003; 127: 432-9.
Gold JS, Antonescu CR, Hajdu C, Ferrona CR, Hussain M, Lewis JJ, et al. Clinicopathologic Correlates of Solitary Fibrous Tumors. Cancer 2002; 94: 1057-68.
Wilson RJ, Gallateau-Salle F, Moran CA. Desmoid tumors of the pleura: a clinicopathologic mimic of localized fibrous tumor. Mod Pathol 1999; 12: 9-14.
![]()