
Vol. 38, n.º 1, 2005
|
REVISTA
ESPAÑOLA DE
Vol. 38, n.º 1, 2005 |
CASUÍSTICA
Juan Segura Sánchez1, Eduardo Solís García1, Antonio Robles Frías1, Jesús Sánchez Sánchez-Vizcaíno2, Vicente Haro Gabalón2
Hospital Infanta Margarita. Avda. de Góngora,
s/n. 14940 Cabra (Córdoba).
1 Servicio de Anatomía Patológica.
2 Servicio de Dermatología.
jsegurasanchez@yahoo.es
RESUMEN
Se presenta un caso de esta infrecuente variedad de dermatofibroma en un varón de 38 años cuya principal característica es la disposición en empalizada de los núcleos. Histopatológicamente, la lesión estaba caracterizada por áreas de empalizada nuclear con formación de cuerpos de tipo Verocay, junto a áreas clásicas de dermatofibroma tipo fibroso. Esta neoplasia debería ser diferenciada de otros tumores benignos y malignos cutáneos con patrón en empalizada. Patólogos y clínicos deberían conocer la existencia de este tipo de dermatofibroma, que no comporta peor pronóstico ni tratamiento adicional.
Palabras claves: dermatofibroma, empalizada, histiocitoma fibroso, cuerpos de Verocay.
SUMMARY
We report a new case of an unusual variant of dermatofibroma of a 38-year-old man in which nuclear palisading is a prominent feature. Histopathologically, the lesions were characterized by areas of nuclear palisading with formation of Verocay-like bodies in addition to the more typical features of the «fibrous» variant of dermatofibroma. This neoplasm should be differentiated from benign and malignant skin tumors with a palisading pattern. Pathologists and clinicians should know of the existence of this type of dermatofibroma and should avoid overdiagnosis and overtreatment.
Key words: dermatofibroma, palisading, fibrous histiocitoma, Verocay bodies.
INTRODUCCIÓN
El dermatofibroma (histiocitoma fibroso benigno), es un tumor dérmico muy frecuente que representa aproximadamente el 3% de los tumores cutáneos (1). Su histogénesis aún en la actualidad sigue siendo controvertida (2), e histológicamente, aunque manteniendo características comunes, se reconocen diferentes subtipos dependiendo de su celularidad y patrones de crecimiento (3).
El dermatofibroma con empalizada, es una variante inusual descrita en 1986 por Schwob y Santa Cruz (4), en una serie de seis casos. Desde entonces, tan sólo, otros dos ejemplos han sido publicados (5,6), si bien uno de ellos era de localización subcutánea (6).
Presentamos un nuevo caso de esta infrecuente variante de dermatofibroma en un hombre de 38 años, haciendo hincapié en su peculiar forma de crecimiento y el diagnóstico diferencial que se plantea.
DESCRIPCIÓN DEL CASO
Hombre de 38 años de edad, sin antecedentes clínicos de interés, con una lesión cutánea polipoide de 1,2 cm, localizada en antebrazo derecho y que refiere desde hace un año. Se extirpa en su totalidad con márgenes de seguridad.
Recibimos un fragmento ovoide de piel de 2,5 ´ 1,5 cm, que muestra una lesión central polipoide, cubierta de piel normal, que mide 1,2 cm y al corte tiene consistencia media y coloración blanquecino-amarillenta de forma difusa.
Histológicamente, se observa una tumoración intradérmica de morfología nodular, parcialmente delimitada aunque no encapsulada, que respeta epidermis, deja una zona «grenz» subepidérmica y no infiltra tejido celular subcutáneo. A diferencia de lo habitualmente observado en los dermatofibroma, no existe inducción epidérmica superficial, estando la mencionada tumoración constituida por un proliferación fusocelular en un estroma fibroso colagenizado con abundantes capilares y, de forma mayoritaria (al menos un 60% de la lesión) las células adoptan un crecimiento «en empalizada», siendo más marcado en la periferia donde adquieren morfología «en collarete» (fig. 1), mientras que en la zona central las empalizadas son más cortas asemejándose a los cuerpos de Verocay de un schwanoma (fig. 2). Asimismo, y de manera dispersa existen áreas de dermatofibroma clásico constituidas por fascículos celulares cortos y arremolinados con patrón estoriforme, células xánticas ocasionales y «atrapamiento» de colágeno periférico (fig. 3). No se observa infiltrado inflamatorio, ni áreas de necrosis y/o hemorragia.
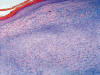
Fig. 1: Collarete
periférico con empalizadas centrales. X10, HE.

Fig. 2: Detalle
de la disposición en empalizada de las células proliferantes. X20, HE.
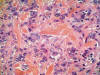
Fig. 3: Crecimiento
celular estoriforme con focales células xánticas. X40, HE.
Desde el punto de vista inmunohistoquímico, las células proliferantes inmunorreaccionan frente a vimentina, factor XIIIa (fig. 4) y en menor cuantía frente a actina de músculo liso y CD68, siendo negativas para S100, CD34, EMA, citoqueratinas y CD117.

Fig. 4: Positividad
de células tumorales para Factor XIIIa. X20, HE.
DISCUSIÓN
Se reconocen múltiples subtipos histológicos de dermatofibroma atendiendo a su celularidad y patrones de crecimiento, entres los cuales podrían reseñarse: el atrófico (7), aneurismático (8), esclerótico (9), de células epitelioides (10), de células claras (11), de células granulares (12), etc.
Clínicamente son tumores dérmicos firmes, no dolorosos, que generalmente no sobrepasan el centímetro de diámetro máximo y que suelen ubicarse en extremidades, principalmente inferiores, siendo más frecuentes en mujeres (1).
Su histogénesis sigue siendo controvertida, discutiéndose si son verdaderos tumores o procesos reactivos; no obstante, la existencia de formas agresivas, recidivantes e incluso con potencial metastático, aparte demostración de su clonalidad, apoyan lo primero (13,14). Durante mucho tiempo se consideró de origen histiocitario, sin embargo hoy parece claro su proceder de fibroblastos.
Histológicamente están caracterizados por una proliferación de fibroblastos, con variable cantidad de colágeno, capilares, células inflamatorias y macrófagos; que, en su forma convencional, no plantean grandes dificultades de diagnóstico, hecho que contrasta con los problemas de diagnóstico diferencial que puede ocasionar el subtipo que presentamos, atendiendo a su rareza y morfología peculiar.
Los dermatofibromas con empalizada, parecen tener predilección por las localizaciones acrales de miembros, no predominan en un sexo o grupo erario determinados y tiene un comportamiento biológico superponible a los dermatofibromas convencionales (4). Desde el punto de vista histológico, son poco celulares, predominando el componente fibroso.
A consecuencia de su distintivo patrón de crecimiento, se plantea el diagnóstico diferencial con otros tumores que, aunque infrecuentemente localizados a nivel intradérmico, también muestran arquitectura con «empalizadas». Entre ellos cabrían destacar: el schwanoma (bien encapsulado e inmunorreaccionan con S100), el perineuroma (con silueta comparable y crecimiento estoriforme, aunque inmunorreactivo frente a EMA y CD34), tumores estromales extragastrointestinales (inmunorreactivos con CD117) y tumores de músculo liso (inmunorreaccionan frente a actina de músculo liso de forma más intensa y difusa, además de hacerlo con desmina y h-caldesmón) (15-18).
En resumen, presentamos el que seria noveno caso, octavo intradérmico, de este subtipo original de dermatofibroma desde que fuese publicado por vez primera en 1986 por Schwob y Santa Cruz (4). Describimos las características histológicas, inmunohistoquímicas y clínicas y mostramos los posibles diagnósticos diferenciales que pueden plantearse debido a su peculiar patrón de crecimiento.
BIBLIOGRAFÍA
Weedon D. Skin Pathology. Tumores y lesiones seudotumorales de los tejidos fibrosos y afines. 1.ª Ed. Londres: Churchill-Livingstone; 2002. p. 759-86.
Nestle FO, Nickoloff BJ, Burg G. Dermatofibroma: an abortive immunoreactive process mediated by dermal dendritic cells? Dermatology 1995; 190: 265-8.
Zelger B, Zelger BG, Burgdorf WH. Dermatofibroma. A Critical Evaluation. Int J Surg Pathol 2004; 12: 333-44.
Schwob VS, Santa Cruz DJ. Palisading cutaneous fibrous histiocytoma. J Cutan Pathol 1986;13: 403-7.
Helm KF, Helm T, Helm F. Palisading cutaneous fibrous histiocytoma. An immunohistochemical study demonstrating differentiation from dermal dendrocytes. Am J Dermatopathol 1993; 15: 559-61.
Fukunaga M. Palisading subcutaneous fibrous histiocytoma. Pathol Int 2004; 54: 360-3.
Beer M, Eckert F, Schmoeckel C. The atrophic dermatofibroma. J Am Acad Dermatol 1991; 25: 1081-2.
Calonje E, Fletcher CD. Aneurysmal benign fibrous histiocytoma: clinicopathological analysis of 40 cases of a tumour frequently misdiagnosed as a vascular neoplasm. Histopathology 1995; 26: 323-31.
Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol 1989; 20: 266-71.
Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol 1989; 120: 185-95.
Zelger BW, Steiner H, Kutzner H. Clear cell dermatofibroma. Case report of an unusual fibrohistiocytic lesion. Am J Surg Pathol 1996; 20: 483-91.
Zelger B, Burgdorf WH. The cutaneous «histiocytoses». Adv Dermatol 2001; 17: 77-114.
Tamada S, Ackerman AB. Dermatofibroma with monster cells. Am J Dermatopathol 1987; 9: 380-7.
Vanni R, Fletcher CD, Sciot R, Dal Cin P, De Wever I, Mandahl N et al. Cytogenetic evidence of clonality in cutaneous benign fibrous histiocytomas: a report of the CHAMP study group. Histopathology 2000; 37: 212-7.
Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). A clinicopathologic and immunohistochemical study. Am J Surg Pathol 1989; 13: 574-80.
Tsang WY, Chan JK, Chow LT, Tse CC. Perineurioma: an uncommon soft tissue neoplasm distinct from localized hypertrophic neuropathy and neurofibroma. Am J Surg Pathol 1992; 16: 756-63.
Yamamoto H, Oda Y, Kawaguchi K, Nakamura N, Takahira T, Tamiya S. c-kit and PDGFRA mutations in extragastrointestinal stromal tumor (gastrointestinal stromal tumor of the soft tissue). Am J Surg Pathol 2004; 28: 479-88.
Bellezza G, Sidoni A, Cavaliere A, Scheibel M, Bucciarelli E. Primary cutaneous leiomyosarcoma: a clinicopathological and immunohistochemical study of 7 cases. Int J Surg Pathol. 2004; 12: 39-44.
![]()