
Vol. 39, n.º 4, 2006
|
REVISTA
ESPAÑOLA DE
Vol. 39, n.º 4, 2006 |
CASUÍSTICA
María Dolores Arias Santos, Marta Pavcovich Ruiz, Miguel Andújar Sánchez, David Martínez Lanao, Laureano León Arencibia
Servicio de Anatomía Patológica. Hospital
Universitario Materno-Infantil de Canarias. Las Palmas de Gran Canaria.
mariadolores.ariassantos@gobiernodecanarias.org
RESUMEN
Introducción: El nefroma mesoblástico congénito (NMC) (hamartoma leiomiomatoso o mesenquimal) es una neoplasia renal congénita. Se diagnostica en los seis primeros meses de vida. Su comportamiento clínico es benigno y el tratamiento es quirúrgico. Paciente y métodos: Describimos el caso de un paciente de una semana de edad que presenta una masa renal. En la ecografía es sólida y sustituye al riñón. El TAC muestra una lesión con márgenes mal delimitados y la estructura heterogénea. Macroscópicamente en la pieza quirúrgica se identificaba una masa intrarrenal bien delimitada, sólida, homogénea, blanquecina y con aspecto fasciculado. Microscópicamente se observa una neoformación compuesta por una población monótona de células fusiformes dispuestas en haces entrelazados. La lesión carece de cápsula y en la interfase con el parénquima renal normal hay células tumorales que rodean a los túbulos y a los glomérulos. Conclusiones: La edad en el momento del diagnóstico y la correcta extirpación del riñón son los dos factores pronósticos más relevantes.
Palabras clave: Nefroma mesoblástico, hamartoma mesenquimal, hamartoma leiomiomatoso, riñón.
SUMMARY
Introduction: Congenital mesoblastic nephroma (CMN) (leiomomatous or mesenchymal hamartoma) is a benign congenital renal neoplasm. This tumor is usually diagnosed along the first six months of life and surgery is current treatment. Patient and methods: A one week of life patient with a renal mass is reported. Ultrasonographically, the mass appearance was solid and replaces the kidney. Computerized tomography (CT) showed a lesion with irregular margins and heterogeneous structure. Grossly, a well delimited intrarenal mass, white, solid, homogeneus and fasciculated was identified. Microscopically, the tumor was composed by a monotonous population of fusiform cells showing an interlacing fasciculated pattern. The mass lacks capsule and the interphase with normal renal parenchyma showed spindle cells surrounding renal tubuli and glomerula. Conclusions: Best prognostic factors are the adequate extirpation and age at the diagnosis.
Key words: Mesoblastic nephroma, mesenchymal hamartoma, leiomiomatous hamartoma, kidney.
INTRODUCCIÓN
El nefroma mesoblástico congénito (NMC), hamartoma renal fetal, hamartoma leiomiomatoso o mesenquimal es un tumor renal raro (1), aunque es el más frecuente en el recién nacido (2), seguido en frecuencia por el tumor de Wilms en el primer año de vida (3), se han publicado casos en adultos (4, 5). La presentación clínica más habitual es la de masa renal detectada en la exploración, también están descritas hematuria, vómitos, hipertensión pulmonar y fallo cardíaco (6, 7). Afecta más frecuente el riñón derecho y todos son unilaterales. El 20% presentan quistes o áreas multiquísticas. Las calcificaciones son raras. El diagnóstico se realiza por pruebas de imagen en los seis primeros meses de vida (la ecografía es característicamente diferente al tumor de Wilms). El tratamiento es la nefrectomía. Aunque se considera una lesión benigna, se han descrito recurrencias locales e incluso metástasis a distancia (pulmón y cerebro) principalmente en aquellos casos que son considerados como NMC atípicos o celulares (con necrosis, hemorragia y/o hipercelulares) (6,8).
Se ha asociado a polihidramnios y a prematuridad (3). También ha sido descrito en relación con hemihipertrofia y síndrome de Wiedeman Beckwith, sugiriendo así una posible relación patogenética con el tumor de Wilms (3).
En la actualidad se considera el tumor renal congénito más frecuente, de comportamiento benigno y pronóstico excelente (7,9).
MATERIAL Y MÉTODOS
Se trata de un paciente de una semana de edad, normosómico y con buen estado general. Presenta una masa renal que en la ecografía es sólida, sustituye el riñón derecho, mide 7 x 4,5 cm y no muestra calcificaciones. En el TAC la lesión tiene márgenes mal delimitados y la estructura es heterogénea. No se evidencian adenopatías, lesiones hepáticas ni nódulos pulmonares.
RESULTADOS
Macroscópicamente la pieza quirúrgica pesaba 98 gramos y medía 6,5 x 5 x 5,3 cm de diámetros mayores. Se identificaba una masa intrarrenal, bien delimitada que, desde el hilio, protruía hacia la pelvis y distorsionaba el sistema pielocalicial, rechazando el parénquima renal residual normal. La tumoración era sólida, homogénea, blanquecina, con aspecto fasciculado, sin quistes, hemorragia ni necrosis. Medía 5,7 x 4,5 x 4,3 cm (fig. 1). Tras la decapsulación afectaba a la grasa perirrenal.
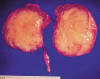
Fig. 1:
Nefroma mesoblástico. Pieza macroscópica. Masa renal bien delimitada, de
consistencia elástica y aspecto fasciculado.
Microscópicamente se observa una masa que infiltra el parénquima renal y forma nódulos separados de la masa principal. Está compuesto por una población monótona de células fusiformes dispuestas en haces entrelazados, con patrón fasciculado y ocasionales remolinos (fig. 2). La interfase entre la masa y el parénquima renal normal no está delimitada por una cápsula, rodeando las células tumorales a túbulos y glomérulos. La densidad celular es moderada, con excepcionales mitosis (fig. 3). Hay mínimos focos de hemorragia. No hay necrosis. Se observan pequeños islotes de cartílago hialino, hematopoyesis extramedular y células que podrían corresponder a rabdomioblastos (fig. 4). No hay infiltración de la pelvis ni del uréter, aunque sí infiltra localmente la grasa perirrenal sin rebasar la cápsula renal.
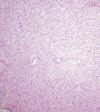
Fig. 2:
Aspecto microscópico mostrando una proliferación monótona de células fusiformes
entrelazadas.
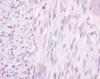
Fig. 3:
Detalle celular con núcleos «blandos», citoplasma eosinófilo y ocasionales
figuras de mitosis.
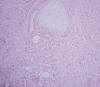
Fig. 4:
Interfase entre lesión y parénquima renal normal, mostrando también un islote de
cartílago hialino.
En el seguimiento superior a un año el niño se encuentra asintomático y libre de enfermedad.
DISCUSIÓN
El NMC produce una masa renal con un incremento en su peso y tamaño, con una media de 130 g (40-700) y de 6 a 8 cm (6,9). Está circunscrito (bien delimitado) y tiene una superficie de corte firme, amarillo-grisácea, arremolinada, con aspecto de leiomioma uterino (2,6). Afecta macroscópicamente al parénquima renal (2).
En el estudio histológico el NMC presenta un patrón de crecimiento fasciculado con haces entrelazados de células fusiformes. Los haces celulares pueden rodear a túbulos y glomérulos, en la periferia tumoral resultado de atrapamiento ya que el tumor carece de cápsula (6). La densidad celular es moderada, sin embargo hay casos denominados NMC atípicos o celulares que pueden tener hipercelularidad, mucha actividad mitótica y focos de necrosis o hemorragia (6).
Este subtipo (NMC atípico o celular) se ha asociado a recurrencia local (7%), invasión del retroperitoneo e incluso metástasis a distancia (pulmón o cerebro) (6,8).
El tratamiento del CMN es quirúrgico (nefrectomía) y no están indicadas ni radioterapia ni quimioterapia. Su comportamiento biológico es benigno en la mayor parte de los casos (2,3,6,7), aunque están descritas en la literatura la recurrencia local, la invasión del retroperitoneo, e incluso las metástasis a distancia (pulmón o cerebro) siempre asociadas a los NMC atípicos o celulares (8).
La mayor parte de las características microscópicas descritas con valor pronóstico en otros tumores, en el CMN carecen de significación. La presencia de focos de cartílago hialino, hematopoyesis extramedular o presencia de rabdomioblastos, así como las mitosis ocasionales o la extensión extrarrenal son datos relativamente frecuentes (en el 20% de los casos) (6). Se cree que la edad en el momento del diagnóstico y la correcta extirpación del riñón son los factores pronóstico más relevantes, de mayor importancia incluso que las características morfológicas (2,9).
Las células que constituyen estos tumores son de características similares a fibroblastos, miofibroblastos o células musculares lisas. Contienen filamentos de vimentina, fibronectina y a veces de actina, y carecen de citoqueratinas. Presenta características de mesénquima secundario, en contraste con el mesénquima mesoblástico o primario, ya que no tiene la capacidad de formar estructuras epiteliales (6). Las anomalías genéticas en el cromosoma 11 que caracterizan al tumor de Wilms (mesoblástico primario) han sido también asociadas al NMC (1,6,10,11).
BIBLIOGRAFÍA
Tomlinson GE, Argyle JC, Velasco S, Nisen PD. Molecular characterization of congenital mesoblastic nephroma and its dintinction from Wilms tumor. Cancer 1992; 70: 2358-61.
Dehner LP, Sibley RK. Kidney. En: Deher, LP, editor. Pediatric Surgical Pathology. Baltimore: Willians & Wilkins; 1987. p. 589-692.
Isaacks H Jr. Tumors. En: Gilbert-Barnes E. editor. Potter´s Pathology of the Fetus and Infant. St. Louis: Mosby; 1996. p. 1242-339.
Tejido Sanchez A, de la Morena Gallego JM, Garcia de la Torre JP, Villacampa Auba F, Martin Muñoz MP, Pamplona Casamayor M et al. Mesoblastic nephroma in the adult: report a new case. Arch Esp Urol 2001; 54: 65-8.
Nakano M, Kawamoto S, Kanimoto Y, Ohkawa Y, Miyazaki Y, Shinmura Y et al. A case of mesoblastic nephroma in an adult patient. Hinyokika Kiyo 2000; 46: 623-6.
Ordoñez NG and Rosai J. Urinary tract. En: Rosai J, editor. Ackerman´s Surgical Pathology. 9th ed. Philadelphia: Ed. Mosby; 2004. p. 1163-360.
Howel CG, Othersen HB, Kiviat NE, Norkool P, Beckwith JB, D´An GJ. Therapy and outcome in 51 children with mesoblastic nephroma: a case report of the National Wilms´Tumor Study. J Pediatr Surg 1982; 17: 826-31.
Heidelberg KP, Ritchey ML, Dausser RC, McKeever PE, Beckwith. Congenital mesoblastic nephroma metastatic to the brain. Cancer 1993; 72: 2499-502.
Pettinato G, Manivel JC, Wick MR, Dehner LP. Classical and cellular (atypical) congenital mesoblastic nephroma: a clinicopathological, ultrastructural, immunohistochemical, and flow cytometric study. Hum Pathol 1989; 20: 682-90.
Roberts P, Lockwood LR, Lewis IJ, Bailey CC, Batcup G, Willians J. Cytogenetic abnormalities in mesoblastic nephroma: a lind to Wilms’tumour? Med Pediatr Oncol 1993; 21: 416-20.
Schofield DE, Yunis EJ, Fletcher JA. Chromosome aberrations in mesoblastic nephroma. Am J Pathol 1993; 143: 714-24.
![]()