
Vol. 39, n.º 4, 2006
|
REVISTA
ESPAÑOLA DE
Vol. 39, n.º 4, 2006 |
CASUÍSTICA
María Macías Díaz1, Nora Sánchez-Mora2, María Cebollero Presmanes2, Gabriel Mandujano Álvarez1, Georgina Velázquez González1, Virgilia Soto Abraham1, Juan Olvera Rabiela1
1 Hospital General de
México.
2 Hospital General Universitario Gregorio Marañón.
RESUMEN
Introducción: La Esclerosis Tuberosa (ET) es un síndrome caracterizado por la formación de hamartomas y tumores en diferentes órganos del cuerpo. La morbimortalidad de esta enfermedad dependerá por lo tanto del órgano involucrado, el tamaño y localización de las lesiones. Paciente y métodos: Presentamos el caso de una mujer de 42 años quien murió por complicaciones de este síndrome Hospital General de México. Se presentan los hallazgos de autopsia.
Palabras clave: Esclerosis tuberosa, hamartomas.
SUMMARY
Introduction: Tuberous Sclerosis is a syndrome characterized by the presence of benign tumors (hamartomas) in multiple organs. Mortality and morbidity keep a close relationship with the location, size, and organs affected by the lesions. Patient and methods: A case of a 40 old women, who died by complications of this syndrome is reported. Autopsy findings are described.
Key words: Tuberous sclerosis, hamartomas.
INTRODUCCIÓN
La esclerosis tuberosa (ET) es un síndrome que se hereda en forma autosómica dominante. Se caracteriza por la formación de hamartomas y tumores benignos en cualquier parte del cuerpo a excepción de músculo esquelético, nervio periférico y médula espinal. La incidencia es de 1 en 6.000 nacidos vivos y un tercio de los casos son esporádicos (1,2).
Se le han detectado mutaciones en 2 genes: TSC 1 y TSC 2. El primero localizado en el cromosoma 9q34, que codifica la proteína hamartina. El segundo se encuentra en el cromosoma 16p13, que codifica la proteína tuberina, cuya mutación se observa en el 80-90% de los casos. Estas dos proteínas en su estado normal funcionan con un complejo que inhibe la proliferación celular (1-4).
Presentamos el caso de una mujer afectada por el síndrome de ET, que desarrollo un cuadro patológico florido en los hallazgos de la autopsia, debido a que alcanzo la edad adulta, lográndose el desarrollo de las múltiples lesiones características de esta patología.
PRESENTACIÓN DEL CASO
Se trata de una mujer de 42 años de edad con diagnostico de esclerosis tuberosa desde la infancia, retraso mental y crisis convulsivas. Historia de carcinoma de endometrio tratado con histerectomía y salpingooferectomía bilateral con tratamiento de quimioterapia 5 años previos a su defunción. Padece insuficiencia renal desde hace 8 años y datos ecográficos de neoplasias renales bilaterales.
Su padecimiento final inició con un cuadro de abdomen agudo, se le realizó laparatomía exploradora, donde se observó trombosis mesentérica y tumores renales bilaterales; por lo que se llevo a cabo resección del yeyuno, íleon, colon derecho y yeyunostomía. Como complicaciones postoperatorias presentó neumonía y peritonitis falleciendo en estas condiciones.
Los hallazgos en el estudio de autopsia fueron los siguientes:
Múltiples hamartomas (tubers) corticales y nódulos subependimarios de astrocitos gigantes en ambos caudados (figs. 1 y 2).
Angiofibromas cutáneos en las extremidades inferiores.
Poliquistosis hepática (fig. 3).
Angiomiolipomas renales bilaterales con absceso izquierdo (fig. 4) con:
- Peritonitis aguda fibrinopurulenta.
- Pericarditis aguda fibrinopurulenta.
Hematoma en la serosa de vesícula biliar.
Absceso pélvico.
Gastritis aguda erosiva.
Neumonía basal.
Bazo accesorio.
Ausencia quirúrgica del útero y anexos bilaterales.
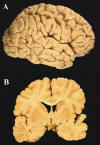
Fig. 1:
A) Hamartomas corticales: se observan ensanchamientos de las circunvoluciones y
aplanamientos corticales que dan un aspecto de tubérculo. B) Al corte en la
corteza hay áreas ensanchadas y mal delimitadas en la materia blanca,
correspondientes a los tubers. Por debajo del epéndimo que rodea al 4.º
ventrículo se observan nódulos de aspecto céreo.
![]()
Fig. 2:
A) Hamartomas subependimarios en el que se
observa una proliferación glial con calcificaciones. B) A mayor aumento se
observa el característico fondo fibrilar y los astrositos gigantes. C) Proteína
ácida fibrilar glial positiva.
![]()
Fig. 3:
A) Poliquistosis Hepática. B) En el corte
histológico observamos que el recubrimiento es por un epitelio cúbico simple.
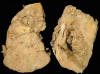
Fig. 4:
Angiomiolipomas bilaterales. En ambos se observa
tejido renal residual en polos superiores. El derecho con formación de absceso
central.
DISCUSIÓN
La esclerosis tuberosa es una enfermedad en la cual los pacientes desarrollan lesiones hamartomatosas en el sistema nervioso central y en otros órganos, debida a un trastorno en la proliferación y diferenciación celular, afectando tejidos tanto de origen neuroectodérmico como mesodérmicos. Se ha observado poca relación entre el fenotipo y el gen expresado (TSC 1 o TSC 2), aunque hay quienes refieren que la tríada clásica (crisis convulsivas, retraso mental, angiofibroma cutáneo) se presenta en los pacientes con genotipo TSC1 (5). Por otra parte se advierte mayor severidad de la enfermedad en quienes expresan TSC2 (6).
La hamartina y la tuberina forman un complejo proteico que suprime una de las vías para la estimulación del crecimiento celular. La mayoría de los proto-oncogenes y genes de supresión tumoral humanos como el RAS y el PTEN, utilizan la vía del complejo Hamartina/Tuberina para regular la proliferación celular (7,8). Aunque los hamartomas generalmente no se desarrollan dentro de los tumores malignos, el reciente descubrimiento de la función de la hamartina y la tuberina en la señalización oncogénica, podría indicar que las mutaciones TSC juegan un rol más importante en la formación de tumores esporádicos en el cáncer de lo anteriormente que se creía (9).
La migración anormal de las neuronas desempeña un papel importante en el desarrollo de las alteraciones neurológicas. Las alteraciones más tempranas que suelen presentarse son el retraso mental, las lesiones cutáneas y las crisis convulsivas que aparecen desde la infancia, mientras que los angiomiolipomas y los quistes renales se diagnostican en edad adulta (10). Esto corresponde a la evolución del caso presentado.
Las lesiones cutáneas son las más frecuentes en la esclerosis tuberosa, y de éstas las manchas café con leche son las más comunes, le siguen los angiofibromas que pueden aparecer en diferentes partes del cuerpo (11).
Las lesiones del sistema nervioso central se presentan en el 80% de los casos, siendo las más frecuentes los tubers corticales y los nódulos subependimarios, también se puede encontrar el astrocitoma de células gigantes, lesión que sólo se ha descrito en este síndrome. La alteración de la arquitectura normal por estas lesiones conduce a retraso mental y crisis convulsivas (12), siendo estas dos alteraciones las primeras causas de morbimortalidad en la ET (13).
Las lesiones renales son la segunda causa de morbimortalidad de la ET y de estas los angiomiolipomas son las más frecuentes. Estos suelen ser de aparición temprana, bilaterales y de mayor tamaño que aquellos que se presentan en pacientes sin ET. Otras de las lesiones renales encontradas son quistes y el carcinoma renal (13).
Los hamartomas hepáticos suelen informarse como hallazgos de autopsia ya que no producen síntomas clínicos ni bioquímicos. En el estudio de Jozwiak y cols., en una serie de de 51 casos de niños con ET, encontró una prevalencia de hamartomas hepáticos en el 23,5% de los casos, los cuales son más frecuentes en mujeres y casi siempre coexisten con angiomiolipomas renales (14).
Además de las lesiones descritas y observadas en este caso, se pueden presentar lesiones pulmonares (linfangioleiomatosis, hiperplasia adenomatosa atípica e hiperplasia micronodular multifocal de neumocitos tipo II), oculares (astrocitoma y manchas hipopigmentadas del iris), quistes óseos, pólipos rectales y hamartomas adrenales (13).
El complejo esclerosis tuberosa continúa siendo una enfermedad de diagnóstico clínico, que se sustenta en la combinación de los criterios mayores y menores consensuados para esta patología (15). El caso que presentamos, es una evidencia más de la amplia variedad de alteraciones presentes un paciente que ha alcanzado la edad adulta y puede desarrollar un cuadro patológico muy florido de esta enfermedad.
BIBLIOGRAFÍA
Jones AC, Shyamsundar MM, Thomas MW, et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 1999; 64: 1305-15.
Cheadle JP, Reeve MP, Sampson JR, et al. Molecular genetic advances in tuberous sclerosis. Hum Genet 2000; 107: 97-114.
Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet 2003; 67: 87-96.
Dabora SL, Sigalas I, Hall F, et al. Comprehensive mutation analysis of TSC1 using two-dimensional DNA electrophoresis with DGGE. Ann Hum Genet 1998; 62 (Pt 6): 491-504.
Dohil MA, Baugh WP and Eichenfield LF. Vascular and pigmented birthmarks. Pediatr Clin North Am 2000; 47: 783-812.
Jones AC, Daniells CE, Snell RG, et al. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum Mol Genet 1997; 6: 2155-61.
Potter CJ, Pedraza LG, Huang H, et al. The tuberous sclerosis complex (TSC) pathway and mechanism of size control. Biochem Soc Trans 2003; 31: 584-6.
Ma L, Chen Z, Erdjument-Bromage H, et al. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005; 121: 179-93.
Knowles MA, Hornigold N and Pitt E. Tuberous sclerosis complex (TSC) gene involvement in sporadic tumours. Biochem Soc Trans 2003; 31: 597-602.
van Slegtenhorst M, Verhoef S, Tempelaars A, et al. Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation. J Med Genet 1999; 36: 285-9.
Jozwiak S, Schwartz RA, Janniger CK, et al. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol 1998; 37: 911-7.
Lendvay TS and Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol 2003; 169: 1635-42.
Crino PB and Henske EP. New developments in the neurobiology of the tuberous sclerosis complex. Neurology 1999; 53: 1384-90.
Jozwiak S, Pedich M, Rajszys P, et al. Incidence of hepatic hamartomas in tuberous sclerosis. Arch Dis Child 1992; 67: 1363-5.
Roach ES, Gomez MR and Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998; 13: 624-8.
![]()