
Vol. 40, n.º 2, 2007
|
REVISTA
ESPAÑOLA DE
Vol. 40, n.º 2, 2007 |
REVISIONES
Samuel Navarro, Antonio Llombart-Bosch, Antonio Pellin, Octavio Burgués, Amparo Ruiz, Marta Piqueras, Rosa Noguera
Departamento de Patología, Facultad de Medicina,
Universidad de Valencia. Blasco Ibáñez, 15. Valencia. 46010 Valencia. España.
samuel.navarro@uv.es
RESUMEN
Antecedentes: Los tumores neuroblásticos son los tumores sólidos extracraneales más frecuentes en la infancia y se caracterizan por una evolución clínica heterogénea que va desde una progresión rápida a una regresión tumoral espontánea. Existen factores pronósticos conocidos que determinan dicha evolución como son la edad, estadiaje, histopatología, estatus de MYCN, ploidía y diversas ganancias y pérdidas cromosómicas. El objetivo del trabajo es describir nuestra experiencia como laboratorio de referencia español para la determinación de estos parámetros pronósticos. Métodos: Material tumoral de pacientes con neuroblastoma, remitido a nuestro laboratorio desde 1992 hasta 2005, ha sido sometido a estudio histopatológico, molecular para determinar la amplificación de MYCN, histoquímico y morfométrico para estudiar la ploidía y se ha introducido la técnica de CGH para el análisis de ganancias y perdidas cromosómicas. Resultados: El seguimiento clínico durante estos años, ha demostrado la importancia pronóstica de la clasificación histológica Internacional Neuroblastoma Pathology Classification (INPC), la relación de un contenido diploide-tetraploide de ADN con el histopronóstico desfavorable, la proporción del 20% de casos con amplificación de MYCN y su carácter pronostico desfavorable, así como la presencia de ganancias y pérdidas cromosómicas como 11q- que confieren mal pronóstico. Conclusiones: Se confirma la necesidad de determinar parámetros morfológicos y genéticos de valor pronóstico con el fin de estratificar la terapéutica apropiada de elección en los pacientes con neuroblastoma.
Palabras clave: Tumores neuroblásticos, pronóstico, histopatología, estatus de MYCN, ploidía.
SUMMARY
Background: Neuroblastic tumors are the most frequent extracranial solid tumors in childhood, and are characterized by a heterogeneous clinic behavior, ranging from a rapid progression of disease to a spontaneous regression. Prognostic indicators that condition such behavior, such as age, staging, histopathology, MYCN oncogene status, ploidy, and diverse chromosomal losses and gains, have been demonstrated. The aim of present work is to describe our experience as reference laboratory for the determination of these prognostic factors in neuroblastic tumors. Methods: Tumor material from patients with neuroblastoma, submitted to our laboratory from 1992 to 2005 has been analyzed. Histopathology following Internacional Neuroblastoma Pathology Classification (INPC) classification, PCR and FISH for MYCN status, static cytometry for ploidy and CGH for chromosomal gains and losses, were performed. Results: The clinical follow-up has demonstrated the prognostic value of INPC, the relationship between diploid-tetraploid DNA content and unfavorable histology, the existence of 20% MYCN amplified cases showing an unfavorable prognosis as well as the presence of chromosomal gains and losses especially 11q-, that confer unfavorable prognosis. Conclusions: We confirm the importance of determining morphological and genetic prognostic parameters in neuroblastic tumors in order to stratify the patients to receive the correct therapy accordingly.
Key words: Neuroblastic tumors, prognosis, histopathology, MYCN status, ploidy.
INTRODUCCIÓN
Los tumores neuroblásticos son los tumores sólidos extracraneales más frecuentes de la infancia y están caracterizados por una evolución clínica heterogénea que abarca desde una progresión maligna rápida a una regresión espontánea. Como resultado, tanto el pronóstico como la respuesta al tratamiento varía ampliamente (1,2). La identificación de los factores pronósticos sólo se puede realizar con garantía si el enfermo ha sido completamente estudiado, para lo que es imprescindible siempre realizar una biopsia, y que ésta sea estudiada en un laboratorio de referencia o con amplia experiencia. Existe una estandarización de criterios clínicos para diagnóstico, estadiaje, y respuesta al tratamiento conocida como Internacional Neuroblastoma Staging System (INSS) (3), definiéndose internacionalmente la necesidad de obtener datos biológicos (histopatológicos y genéticos) para crear nuevos grupos terapéuticos. Los análisis histopatológicos tienen un importante valor para conocer la biología tumoral (4-6). En 1984, Shimada y colaboradores (7) describieron un sistema de clasificación basado en la cantidad del estroma schwanniano, el grado de diferenciación, el índice de mitosis-cariorrexis (MKI) y la edad al diagnóstico, distinguiendo dos grupos de histopronóstico: favorable y desfavorable. Actualmente se utiliza la Internacional Neuroblastoma Pathology Classification (INPC) (8,9), basada en la clasificación original de Shimada con pequeñas modificaciones. Las anormalidades genéticas que caracterizan estos tumores son conocidas desde hace tiempo y algunas de ellas poseen claro valor pronóstico (10-14). Entre los marcadores genéticos de neuroblastomas (NB) agresivos se incluyen la diploidía o tetraploidía (15,16), la amplificación del proto-oncogén MYCN (17-19), la deleción del 1p (20-22), y la ganancia del 17q (23-26). Esta última reestructuración, que se presenta con una alta frecuencia, puede resultar de una traslocación no balanceada del 17q con más de 20 regiones cromosómicas diferentes (27,28). Recientemente, se ha encontrado que los tumores con disomía del cromosoma 17 y un presumible estadio favorable puede representar un subgrupo de riesgo alto (29). Además, se han descrito como marcadores de posible valor pronóstico las pérdidas frecuentes de heterocigosidad (LOH) en otras regiones cromosómicas, especialmente 2q (30%), 3p (15.3%), 4p (19.5%), 9p (36%), 11q (5-44%), 14q (18-23%) y 18q (31%) (30-34). Se han descrito asociaciones entre pérdidas de 11q, 3p y 14q, que se correlacionan inversamente con la amplificación de MYCN y la deleción de 1p, identificando subgrupos diferentes de tumores avanzados (30-33).
De la multitud de factores pronósticos conocidos, muchos de ellos no tienen un valor independiente y se presentan asociados con otros de mayor peso específico. La edad, el estadio y la amplificación del MYCN son los factores admitidos por todos los grupos cooperativos como marcadores con valor pronóstico independiente. A ellos se añaden, con menor valor, la clasificación histológica de Shimada, la deleción del 1p y la ploidía. Otros marcadores biológicos como el TRKA/B, el MDR1, la ganancia de 17q, y otras LOH han demostrado valor pronóstico en estudios aislados, pero no han sido utilizados todavía en grandes estudios cooperativos para conocer cuál es su valor pronóstico real (35-37).
Los enfermos se clasifican, por tanto, en grupos de riesgo con recomendaciones terapéuticas diferentes para cada uno de ellos, existiendo una concordancia del 85% entre las clasificaciones utilizadas por los mayores grupos cooperativos en NB. El grupo de alto riesgo (HR) es el más numeroso (45%), compuesto principalmente por pacientes mayores de 1 año con estadio 4, pero también incluye a cualquier otro estadio de la enfermedad, excluyendo el estadio 1 con amplificación del MYCN, independientemente de la edad (grupo de NB Sociedad Internacional de Oncología Pediátrica [SIOP]-Europa). El pronóstico de estos enfermos es malo, incluso con tratamientos muy agresivos. Sólo la amplificación del MYCN confiere un valor pronóstico negativo a los tumores que la presentan (38-40).
El objetivo fundamental del presente trabajo es describir la experiencia de 14 años como Centro Nacional de Referencia para estudios biopatológicos del neuroblastoma en relación al valor pronóstico de los factores anteriormente mencionados, con especial énfasis en la clasificación histopronóstica INPC, el estatus del gen NMYC y su proteína codificada, la ploidía tumoral y el estudio de diversas ganancias y pérdidas cromosómicas.
MATERIAL Y MÉTODOS
Pacientes
El material tumoral de los pacientes estudiados fue remitido entre Enero de 1992 y Junio 2005 al Centro de Referencia Nacional de Estudios Anatomopatológicos y Biológicos de Neuroblastoma de la SEOP (Sociedad Española de Oncología Pediátrica) (Dpto. Patología, Universidad de Valencia). El diagnóstico de NB y estadiaje fue realizado según la INSS.
Histopatología e inmunohistoquímica
El diagnóstico histológico, basado en las recomendaciones de la INPC fue revisado por dos patólogos del Centro de Referencia Nacional. Fueron analizados los siguientes parámetros morfológicos: porcentaje de estroma schwanniano, MKI (bajo, medio y alto siendo <100, 100 a 200 y >200 mitosis o cariorrexis por 5.000 células tumorales respectivamente), grado de diferenciación basado en la presencia o ausencia de neuropilo, diferenciación gangliónica (indiferenciado, pobremente diferenciado o en diferenciación), y la presencia o ausencia de calcificación.
Los casos analizados fueron clasificados en subgrupos de histopronóstico favorable y desfavorable dependiendo de la categoría y subtipo histológico, del índice mitosis-cariorrexis y de la edad.
El diagnóstico de la categoría de neuroblastoma indiferenciado requirió confirmación inmunohistoquímica con la detección del marcador NB84.
Se elaboró una matriz tisular con 49 casos de neuroblastoma con amplificación y ganancia de NMYC, utilizando un sistema manual Beecher MTA.
El estudio inmunohistoquímico de la proteína Nmyc se realizó utilizando el anticuerpo monoclonal antiMYCN protein clon 8.4B suministrado por BD Pharmingen y un sistema de detección ENVISION (Dako Cytomation).
Análisis genéticos
Los tumores fueron analizados genéticamente usando la técnica de citometría estática, de FISH y de CGH. Para ello se realizaron secciones de las piezas incluidas en parafina, improntas celulares y se extrajo ADN de la misma pieza tumoral utilizada para el estudio histopatológico. Para interpretar los resultados de todas estas técnicas genéticas fue imprescindible conocer el porcentaje de células neuroblásticas en las secciones tumorales.
Para la medición del índice de ADN por citometría estática se utilizaron secciones de material incluido en parafina de 6 micras de grosor teñidas con el reactivo de Feulgen (hidrólisis ácida con ácido clorhídrico 30-50 minutos y Schiff 1 hora) y el sistema de análisis de imagen Olympus CUE-2. Se midió la densidad óptica integrada de 250-500 núcleos de células tumorales y 25-50 núcleos linfocitarios (referencia interna) por caso y los resultados se basaron en el Consenso Europeo (41).
Las improntas celulares con más de 200 células neuroblásticas fueron utilizadas para detectar amplificación de MYCN y para conocer la deleción de la región 1p36 por FISH. Las sondas utilizadas y las condiciones de hibridación, lavados, detección han sido previamente descritas (42,43). Brevemente, se utilizaron sondas Q-BIOgene N-myc (2p24)/ 2 a-satélite y 1p36 midisatélite/ 1 alfasatélite de doble color. La hibridación se realizó en cámara húmeda a 37° C durante 16 horas. Los lavados posteriores se hicieron con 50% formamida/2X SSC y 2X SSC. Las preparaciones fueron contrateñidas con Dapi/antifade. Se analizaron de 200 a 500 núcleos por caso con un microscopio epifluorescente Zeiss axioplan2. La amplificación de MYCN fue considerada cuando existía incrementado más de 4 veces el número de señales de N-Myc en relación con el número de señales de cromosoma 2 alfa-satélite. La ganancia de MYCN fue reconocida cuando aparecía de 1 a 4 veces incrementado el número de señales de N-Myc en relación con el número de señales de cromosoma 2 alfa-satélite. La amplificación de NMYC también se estudio con PCR diferencial con el gen de la beta globina.
Para la detección de secuencias de ADN NMYC en suero se extrajo ADN a partir de 200 microlitros de plasma o suero utilizando el QI Ampl. Blood KIT (Quiagen), seguido de amplificación por PCR y Real Time PCR cuantitativa usando el sistema Taqman detection y el ABI Prism 7700 sequence detection system (Applied Biosystems), utilizando como gen de referencia el ProIL1beta localizado en 2q13-24.
La deleción de 1p36 fue detectada por la presencia única de una región subtelomérica del brazo corto del cromosoma 1 (p36 midisatélite). Consideramos como desequilibrio de 1p36 cuando existía una desproporción entre el número de centrómeros del cromosoma 1 y la región 1p36, siempre con la existencia de más de una región subtelomérica del brazo corto del cromosoma 1.
Utilizamos la técnica de CGH descrita por Kallioniemi y colaboradores (44) con algunas modificaciones. Brevemente, se utilizaron metafases (46XY) (Vysis). EL ADN tumoral fue marcado con fluoresceína-12-dUTP (Vector), y el ADN de referencia comercial y previamente marcado con Spectrum red (Vysis). El tamaño de los fragmentos finales fue de 500-2.000 bp. Se mezcló la misma cantidad (300ng) de ADN tumoral y de ADN de referencia precipitándose con 20 mg de ADN Cot-1 (Roche). Los distintos ADNs fueron disueltos en 50% formamida/2X SSC y desnaturalizados 5-7 minutos a 75° C. Las preparaciones de metafases fueron desnaturalizadas en 70% formamida/2X SSC, 3-5 minutos a 73° C y deshidratadas en series de etanol. La co-hibridación de los ADNs sobre las metafases normales se realizó en cámara húmeda durante 48-72 horas. Los lavados posteriores se realizaron con 50% formamida/2X SSC y 2X SSC a 45° C, seguidos de 0.2X SSC y tampón fosfato. Las preparaciones fueron contrateñidas con Dapi/antifade. Se analizaron entre 10 y 20 metafases por caso con un microscopio epifluorescente Zeiss axioplan2 con video cámara CCD S31. El análisis de las imágenes se hizo usando el software ISIS v.3.1. Consideramos los ratios verde/rojo >1,2 y >1,5 como número de copias de ADN con ganancia y amplificación, respectivamente, y ratios <0,8 como perdidas del número de copias de ADN.
RESULTADOS
Estudio histológico
El estudio histopatológico siguiendo la clasificación INPC, fue aplicado en dos trabajos que han sido recientemente publicados (45,46)
En un primer estudio, se analizaron 256 casos que fueron clasificados en subgrupos favorable (88 casos) y desfavorable (168 casos). La distribución según el subtipo histológico demostró que la mayoría eran neuroblastomas pobremente diferenciados (63%). El seguimiento clínico demostró que los neuroblastomas indiferenciados presentaban una tasa de mortalidad alta (62.9%) en comparación con la supervivencia global del 91,7% de los neuroblastomas en diferenciación (figs. 1 a y b).
Uno de los factores morfológicos con impacto pronóstico fue el índice mitosis-cariorrexis (MKI), con una tasa de supervivencia baja (47,7%) en aquellos casos con MKI alto si se compara con supervivencia del 87,9% en casos con MKI bajo.
Finalmente, en este primer estudio, el subgrupo de casos con histopronóstico desfavorable correlacionó con una supervivencia global del 59,2% con respecto al 93.8% de los casos considerados favorables.
Otro de los estudios realizados se refiere al valor histopronóstico de la clasificación INPC en neuroblastomas localizados especialmente en estadio 2. Para ello hemos colaborado en un protocolo multicéntrico Europeo denominado LNESG1, en el cual, los 124 pacientes incluidos solo recibieron un tratamiento quirúrgico. La histopatología de dichos tumores primarios fue revisada por el panel de patólogos de la SIOP. La clasificación INPC pudo aplicarse en 115 casos con un total de 91 casos favorables (73,0%) y 24 casos desfavorables (19,4%). Tras un seguimiento de 5 años, tanto la supervivencia global como el intervalo libre de recaídas se asociaron al histopronóstico favorable o desfavorable con carácter estadísticamente significativo.
Estudio del estatus de NMYC
El análisis de la amplificación génica del NMYC lo enfocamos desde tres puntos de vista:
Estudio de NMYC por FISH y PCR: En una primera etapa (1992-1999), sobre un estudio de 264 casos, tan solo demostramos amplificación en el 13% de los neuroblastomas, los cuales estuvieron correlacionados con histopronóstico desfavorable. Comparativamente, el estudio actualizado (2000-2005), que incluye los resultados por FISH, revela un porcentaje mayor (20%) de casos con NMYC amplificado y que también se asocian a un histopronóstico desfavorable. Además, hemos tenido ocasión de analizar alteraciones poco frecuentes como la amplificación focal, heterogénea y la ganancia de NMYC, definida por FISH como un aumento de 2-4 señales con respecto al numero de centrómeros del cromosoma 2 y cuyo valor pronóstico y su influencia en el diseño de la estrategia terapéutica están en controversia (figs. 1 e y f).
Estudio de amplificación y expresión de Nmyc con matrices titulares: En nuestra matriz tisular de 50 casos, hemos detectado amplificación en 41 casos, que a su vez sobreexpresan inmunohistoquímicamente la proteína Nmyc (figs. 1 c y d). Sin embargo no hemos detectado en la matriz los casos de ganancia que habían sido detectados previamente por FISH en las improntas citológicas de los mismos casos.
Estudio de secuencias de NMYC en suero: Hemos observado en los pacientes analizados una gran sensibilidad de la técnica puesto que de 33 pacientes, 6 de los cuales con tumores amplificados, hemos detectado 5 con ADN NMYC circulante, mientras que en ninguno de los pacientes no amplificados se detectaron secuencias de ADN NMYC en suero.
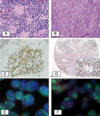
Fig. 1:
(A y B) Histopatología de Neuroblastoma
pobremente diferenciado y en diferenciación. (C y D) Expresión
Inmunohistoquímica nuclear de proteína MYCN en una sección completa y en una
matriz tisular. (E y F) Amplificación de MYCN con técnica de FISH en forma de
dobles minutos y HSR.
Estudio de la ploidia tumoral
El análisis del contenido tumoral de ADN efectuado con citometría estática mostró en un primer estudio (1992-1999) un porcentaje alto de casos aneuploides (40%), mientras que en el estudio actual (2000-2005), estos casos se han agrupado en el rango triploide, con un total de 63% comparativamente con un 26% de casos diploides y un 11% tetraploides. Tanto en un estudio como en otro, la triploidia se asoció a un histopronóstico favorable, mientras que los rangos diploides y tetraploide correlacionaron con histopronóstico desfavorable.
Estudio de ganancias y pérdidas cromosómicas
El estudio de la integridad del brazo corto del cromosoma 1 (del1p), mostró en un primer análisis un porcentaje del 24% de casos, que ha aumentado al 30% en el estudio actualizado (2000-2005). En ambos periodos, su presencia se asoció a histopronóstico desfavorable.
Otras ganancias y pérdidas cromosómicas estudiadas se refieren a la ganancia del 17q observada en 30 de 48 casos, así como las pérdidas de 11q (11 de 35 casos) y la pérdida de 14q (observada en 4 casos de 28 analizados).
Finalmente hemos tenido la oportunidad de estudiar 60 casos del protocolo Europeo de alto riesgo, tratando de analizar comparativamente las ganancias y pérdidas cromosómicas detectadas por FISH con el análisis genómico completo con CGH.
El análisis por FISH del oncogén MYCN y de la integridad de la región 1p36 en los 60 casos estudiados permite dividir la serie tumoral en tres grupos. Grupo 1: amplificación del MYCN, con deleción de 1p36 (n=29) y no deleción de 1p36 (n=2); grupo 2: no amplificación del MYCN con deleción de 1p36 (n=7) y no deleción de 1p36 (n=14); grupo 3: ganancia del MYCN con deleción de 1p36 (n=6), no deleción de 1p36 (n=1) y desequilibrio de 1p36 (n=1). Los datos clínicos, histopatológicos y de ploidía relacionados con dichos grupos están resumidos en la tabla 1.
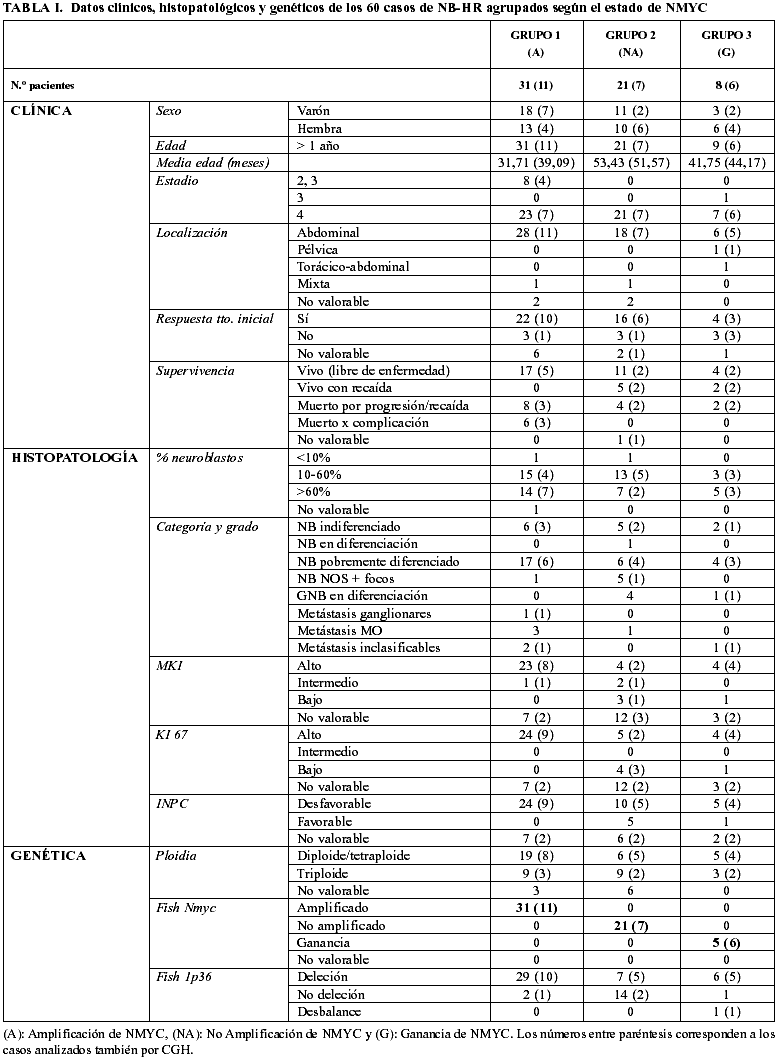
Todas las anomalías genéticas diagnosticadas por FISH son confirmadas en los 24 casos analizados con la técnica de CGH. La mayor parte de los casos (14/24) presentan asociados amplificación o ganancia de MYCN, deleción de 1p36 y ganancia del brazo q del cromosoma 17 o presencia de cromosoma 17 en disomía (fig. 2). En la tabla 2 se presenta la asociación de los grupos 1, 2 y 3 con los resultados de ploidía, número de desequilibrios cromosómicos y alteraciones cromosómicas presentes, descritas en la literatura como marcadores de posible valor pronóstico), INPC y las características clínicas.
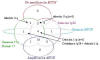
Fig. 2:
Relaciones entre MYCN, 1p36, cromosoma 17 y 11q en los 24 casos analizados por
CGH.
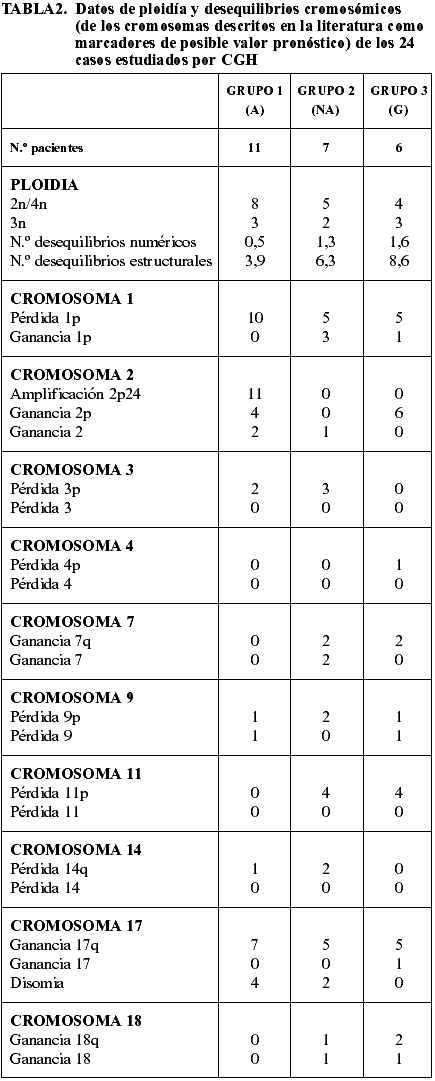
La amplificación del MYCN (tabla 2, figs. 3 y 4) presente en 11 casos se asocia a deleción de 1p36 (10/11) y ganancia del brazo largo del cromosoma 17 (7/11) o cromosoma 17 en disomía (4/11). La amplificación de MYCN (región 2p24) está asociada a ganancia de otras regiones del brazo corto del cromosoma 2 (p21-23) o ganancia completa del cromosoma 2. La deleción de 1p se limita a la región 1p36 en un caso siendo en el resto de casos la deleción del brazo corto del cromosoma 1 mayor; en tres casos la deleción del brazo 1p es completa. Únicamente en un caso el cromosoma 1 está sin deleción. Estos casos MYCN amplificados (11/24) presentan pocas ganancias y pérdidas completas cromosómicas y un número escaso de reestructuraciones parciales del resto de cromosomas (media de 3,9 reestructuraciones por tumor). Un caso presenta deleción de 3p22 y en otro caso se observa una deleción de 14q31 estando ausente en todos los casos la deleción del brazo del cromosoma 11q. Ocho de estos casos son diploides y tres triploides. Todos los casos siguiendo la INPC presentan un histopronóstico desfavorable y todos excepto uno presentan un alto MKI junto con una alta expresión de Ki67. La edad media al diagnóstico es de 39,09 meses. A pesar de que todos los enfermos excepto 1 respondieron inicialmente al tratamiento, fallecieron 5 enfermos por progresión y uno por complicaciones.

Fig. 3:
Resumen de perfiles de CGH de amplificaciones (barras verde gruesas), ganancias
(barras verde finas), y pérdidas (barras rojas) en el grupo de los casos con
amplificación de NMYC.

Fig. 4:
Perfil de CGH de un caso amplificado.
La no amplificación del MYCN (tabla 2, figs. 5 y 6) presente en 7 casos se asocia a deleción de 1p36 y a la ganancia del brazo q del cromosoma 17 en 5/24 casos. La deleción de 1p se limita en todos los casos a la región 1p36 asociándose en tres casos a ganancias parciales de regiones en 1q, un caso presenta una deleción intersticial 1p33-35 y otro caso presenta el brazo 1p íntegro. Las ganancias y pérdidas cromosómicas parciales son frecuentes en estos casos sin amplificación de MYCN (media de 6,3 reestructuraciones por tumor), entre ellas la deleción del 11q14-23 está presente en 4 casos, en dos de ellos asociada a deleción de 3p21 y en los otros dos casos asociada a deleción 14q31. Un caso presenta deleción de 3p21 sin deleción de 11q. Cinco de estos casos son diploides/tetraploides y dos triploides (ambos casos presentan deleción de 11q). El histopronóstico en los cuatro casos valorables es desfavorable aunque únicamente dos casos presentan un alto MKI junto con una alta expresión de Ki67. La edad media al diagnóstico es de 51,57 meses. Todos los enfermos excepto 1 respondieron inicialmente al tratamiento falleciendo 2 enfermos por progresión. Dos casos se encuentran en progresión de su enfermedad.

Fig. 5:
Resumen de perfiles de CGH de casos no amplificados (barras verde finas), y
pérdidas (barras rojas) en el grupo de los casos sin amplificación de NMYC.

Fig. 6:
Perfil de CGH de un caso no amplificado.
Por último, la ganancia del MYCN (tabla 2, figs. 7 y 8) presente en 6 casos, se asocia a deleción de 1p36 y a ganancia del brazo del cromosoma 17q en 5 casos; un caso no presenta deleción de 1p36 sino desequilibrio de 1p36, estando en otro caso presente una ganancia completa del cromosoma 17. Cuatro casos presentan deleciones en 11q sin estar asociada a deleción de 3p ni deleción de 14q. Las ganancias y pérdidas cromosómicas parciales son frecuentes en todos los casos con ganancia de MYCN (media de 8,6 reestructuraciones por tumor). Cuatro de estos casos son diploides/tetraploides y dos triploides (uno de estos casos presenta deleción de 11q). El histopronóstico en los cuatro casos valorables es desfavorable presentando un alto MKI junto con una alta expresión de Ki67. La edad media al diagnóstico se encuentra en 44,17 meses. Tres casos no responden al tratamiento, encontrándose en progresión de su enfermedad junto con un cuarto enfermo que tras responder inicialmente al tratamiento se encuentra en la misma situación.

Fig. 7:
Resumen de perfiles de CGH de casos con ganancia (barras verde finas), y
pérdidas (barras rojas) en el grupo de los casos sin amplificación de NMYC.

Fig. 8:
Perfil de CGH de un caso con ganancia.
DISCUSIÓN
La heterogeneidad clínica observada en los tumores neuroblásticos se asocia a múltiples factores biológicos, fenotípicos y genotípicos cuyo análisis permite obtener datos de importancia pronóstica y que condicionan las diferentes estrategias terapéuticas a usar en cada paciente.
Clásicamente, existen parámetros clínicos de demostrado valor pronóstico como son la edad del niño y el estadio tumoral localizado o avanzado en el momento del diagnóstico de la enfermedad neoplásica.
La tipificación histológica de las categorías tumorales en base a criterios morfológicos como la diferenciación y el índice mitosis cariorrexis, ha sido demostrada por Shimada y cols y Joshi y cols (4-9). En nuestro trabajo confirmamos el valor histopronóstico de la clasificación INPC propuesta por Shimada y más aún demostramos que al menos en los neuroblastomas localizados es el marcador pronóstico más importante y que va a condicionar la estrategia terapéutica en los casos de recidiva en los neuroblastomas localizados (Protocolo Europeo LNESG2, en curso).
La amplificación del oncogén NMYC es otro de los parámetros biológicos asociados a progresión de enfermedad, estadios avanzados y por tanto pronóstico desfavorable. Su determinación por métodos de genética molecular es necesaria en todos los protocolos cooperativos de tratamiento oncológico. En nuestro trabajo hemos confirmado la frecuencia en que aparece esta alteración genética (20%) y su asociación a otros factores de pronóstico desfavorable. Un dato interesante que hemos estudiado es la detección de secuencias de ADN NMYC en el suero de pacientes con amplificación así como el valor de su detección para predecir con antelación las recidivas en estos pacientes (47).
El valor pronóstico de la detección del contenido de ADN o ploidía ha sido demostrado en niños menores de un año (16). En nuestro trabajo, hemos observado una relación entre los casos triploides y el histopronóstico favorable, comparado con casos diploides y tetraploides que presentan una evolución desfavorable.
La clasificación de riesgo dentro del NB se apoya actualmente en la histopatología y en la determinación de escasos factores clínicos y biológicos (1,34). A pesar de que la estratificación en base a estos riesgos ha permitido una marcada mejora terapéutica, los datos sugieren un hipotético modelo donde el estadio final del NB es predeterminado en el momento que adquiere unos cambios genéticos específicos, por lo que otro objetivo principal en nuestro estudio es la predicción más precisa del comportamiento de la enfermedad y de la valoración del riesgo de cada paciente al diagnóstico en base a cambios genéticos específicos, en el grupo de tumores neuroblásticos desfavorables de estadio avanzado (NB-HR).
Utilizando la técnica del FISH y de citometría estática hemos observado, en consonancia con estudios previos, que los 60 tumores neuroblásticos desfavorables de estadio avanzado recibidos en un periodo de tres años en el Centro de Referencia Nacional, son tumores en progresión con características genéticas heterogéneas. Por este motivo, nos planteamos aplicar la CGH metafásica, a un pequeño grupo de estos tumores para valorar la posibilidad de detectar nuevas diferencias genéticas, definir subtipos tumorales basados en estratificación genética y por tanto aplicar al diagnóstico de forma amplia esta técnica en el Centro de Referencia.
Con el método de CGH se obtiene una visión global o perfil genómico de la heterogeneidad genética y de las interrelaciones de sus desequilibrios (48,49). Con el uso del análisis de CGH metafásico se detectan nuevas alteraciones genéticas, a pesar de que esta técnica tiene un límite de resolución de 10-20 Mb (50-53). Subdividiendo los tumores únicamente según el estatus de MYCN, observamos que la ganancia del brazo largo del cromosoma 17 es la anomalía más frecuente compartida por los tres subgrupos seguida por la deleción de 1p36. Recientemente Vandesompele y cols. (29) han detectado que en los tumores neuroblásticos favorables, la presencia del cromosoma 17 en disomía representa un signo de alto riesgo equiparable a la ganancia de 17q; por esta razón hemos englobado este dato en nuestro estudio junto con la ganancia de 17q. Estos hallazgos han sido descritos con alta frecuencia en los NB-HR pero no han sido utilizados todavía en grandes estudios cooperativos para conocer cuál es su valor pronóstico real (27,28). Asimismo, en los grupos de MYCN no amplificado y ganancia de MYCN hemos observado tumores que presentan en común la pérdida del brazo largo del cromosoma 11, hallazgo no presente en el grupo de tumores con amplificación de MYCN. Debido a que los estudios realizados mediante las técnicas de CGH metafásica y micro-matrices de CGH con BAC, cDNA u oligonucleótidos que pueden detectar alteraciones genéticas y de expresión con una mayor resolución, están permitiendo la identificación en NB de patrones de expresión génica correlacionadas con desequilibrios genéticos específicos, en particular con la pérdida del cromosoma 11q (51-54), consideramos esta alteración como identificativa de un subgrupo tumoral. Así, en nuestro estudio podemos diferenciar dos grupos genéticos claros: el grupo 1 de tumores MYCN amplificados y el grupo 2 de tumores con del 11q.
El grupo 1 lo identificamos y definimos por la presencia de amplificación de MYCN no habiendo otros hallazgos genéticos específicos. Estos casos presentan muy pocas alteraciones genómicas, indicando que la amplificación de MYCN es suficiente para determinar en estos tumores un fenotipo agresivo, aunque ocurren otros cambios genéticos, incluidos la pérdida de 1p36 y la ganancia del 17q. La deleción 1p asociada a la amplificación de MYCN afecta, en los diferentes tumores, a regiones amplias de dicho brazo cromosómico teniendo como consecuencia la pérdida de múltiples genes. Se observa así mismo la integridad del brazo largo del cromosoma 11. Dentro de los NB-HR los pacientes de este grupo presentaban una media de edad al diagnóstico menor (39,09 meses) y progresan rápidamente.
El grupo 2 presenta mayor número de anormalidades estructurales, estando presente la deleción 11q. Estos casos están incluidos tanto en el grupo MYCN no amplificado como en el grupo de ganancia de MYCN. Se han identificado genes cuyos patrones de expresión se correlacionan con la pérdida de 11q, apoyando el concepto de que la pérdida del 11q representa a un subtipo genético distinto dentro de los neuroblastomas. Por tanto consideramos que los tumores con la pérdida del cromosoma 11q forman un subgrupo diferenciado de tumores que a su vez pueden o no presentar asociada la ganancia del MYCN, pérdida del 1p, 3p o del 14q. Los tumores incluidos en este subgrupo crecen más despacio, siendo la media de edad al diagnóstico mayor (52 meses) y por tanto acumulan mayor número de anormalidades genéticas secundarias, presumiblemente por el tiempo más largo de evolución.
El grupo de tumores que no presentan amplificación de MYCN ni deleción del 11q sigue siendo un grupo genéticamente heterogéneo en donde no hemos podido detectar con la técnica de CGH metafásica anomalías comunes identificativas aunque sí están presentes la ganancia del 17q y/o el cromosoma 17 en disomía.
CONCLUSIONES
Concluimos que el estudio histopatológico siguiendo la clasificación INPC es un factor de primer orden no solo necesario para el diagnóstico sino también para la estratificación terapéutica, especialmente en los casos de neuroblastoma localizado (56-57).
El estatus de NMYC debe ser determinado en todos los casos, pues condiciona la decisión terapéutica. La metodología recomendada en todos los protocolos cooperativos multinacionales es el FISH.
El estudio de la ploidía por citometría estática aporta información relacionada con el histopronóstico favorable o desfavorable.
El estudio genómico de ganancias y pérdidas cromosómicas aporta información pronóstica y abre nuevas vías para el estudio de la génesis y progresión tumoral de los tumores neuroblásticos.
Como conclusión final, nuestro grupo ha identificado, utilizando la CGH metafásica, dos grupos genéticos diferenciados que pueden tener implicaciones diagnósticas y terapéuticas importantes. A pesar de tratarse de un reducido número de casos analizados y con un corto periodo de seguimiento, ha sido posible definir cambios genéticos asociados a los distintos subgrupos de pacientes. Estos resultados ponen de manifiesto la necesidad de aplicar estas técnicas como complemento a las pruebas diagnósticas actuales.
AGRADECIMIENTOS
A los Profesores A. Pellin, R. Noguera, A. Ruiz, Dra D. Sanchez, Lda Marta Piqueras, D. Alejo Sempere y Dña Cristina Mongort del Departamento de Patología de la Universidad de Valencia por su colaboración directa en la realización del presente trabajo de investigación.
Trabajo financiado por el proyecto «Patología Molecular de los tumores sólidos infantiles: aportación al diagnóstico y a la individualización del tratamiento». G03/089 del Instituto Carlos III (Madrid) FIS PI 05/0383 y FIS PI 06/1576.
BIBLIOGRAFÍA
Cotterill SJ, Pearson AD, Pritchard J, Foot AB, Roald B, Kohler JA, Imeson J. Clinical prognostic factors in 1277 patients with neuroblastoma: results of The European Neuroblastoma Study Group «Survey» 1982-1992. Eur J Cancer 2000; 36: 901-8.
Evans AE, Silber JH, Shpilsky A, D’Angio GJ. Successful management of low-stage neuroblastoma without adjuvant therapies: a comparison of two decades, 1972 through 1981 and 1982 through 1992, in a single institution. J Clin Oncol 1996; 14: 2504-10.
Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 1993; 11: 1466-77.
Chatten J, Shimada H, Sather HN, Wong KY, Siegel SE, Hammond GD. Prognostic value of histopathology in advanced neuroblastoma: a report from the Childrens Cancer Study Group. Hum Pathol 1988; 19: 1187-98.
Joshi VV, Rao PV, Cantor AB, Altshuler G, Shuster JJ, Castleberry RP. Modified histologic grading of neuroblastomas by replacement of mitotic rate with mitosis karyorrhexis index. A clinicopathologic study of 223 cases from the Pediatric Oncology Group. Cancer 1996; 77: 1582-8.
Shimada H, Stram DO, Chatten J, Joshi VV, Hachitanda Y, Brodeur GM, Lukens JN, Matthay KK, Seeger RC. Identification of subsets of neuroblastomas by combined histopathologic and N-myc analysis. J Natl Cancer Inst 1995; 87: 1470-6.
Shimada H, Chatten J, Newton W.A, Jr Sachs, N Hamoudi, A.B Chiba, T Marsden, H.B Misugi, K. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J Natl Cancer Inst 1984; 73: 405-16.
Shimada H, Umehara S, Monobe Y, Hachitanda Y, Nakagawa A, Goto S, Gerbing RB, Stram DO, Lukens JN, Matthay KK. International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 2001; 92: 2451-61.
Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 1999; 86: 364-72.
Cox D, Yuncken C, Spriggs AI. Minute Chromatin Bodies in Malignant Tumours of Childhood. Lancet 1965; 62: 55-8.
Brodeur GM, Sekhon G, Goldstein MN. Chromosomal aberrations in human neuroblastomas. Cancer 1977; 40: 2256-63.
Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, Brodeur G, Goldstein M, Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983; 305: 245-8.
Gilbert, F, Feder, M, Balaban, G, Brangman, D, Lurie, DK, Podolsky, R, Rinaldt, V, Vinikoor, N, Weisband, J. Human neuroblastomas and abnormalities of chromosomes 1 and 17. Cancer Res 1984; 44: 5444-9.
Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol 1999; 17: 2264-79.
Hayashi Y, Habu Y, Fujii Y, Hanada R, Yamamoto K. Chromosome abnormalities in neuroblastomas found by VMA mass screening. Cancer Genet Cytogenet 1986; 22: 363-4.
Look AT, Hayes FA, Nitschke R, McWilliams NB, Green AA. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N Engl J Med 1984; 311: 231-5.
Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984; 224: 1121-4.
Tsuda T, Obara M, Hirano H, Gotoh S, Kubomura S, Higashi K, Kuroiwa A, Nakagawara A, Nagahara N, Shimizu K. Analysis of N-myc amplification in relation to disease stage and histologic types in human neuroblastomas. Cancer 1987; 60: 820-6.
Christiansen H, Sahin K, Berthold F, Hero B, Terpe HJ, Lampert F. Comparison of DNA aneuploidy, chromosome 1 abnormalities, MYCN amplification and CD44 expression as prognostic factors in neuroblastoma. Eur J Cancer 1995; 31A: 541-4.
Christiansen H, Lampert F. Tumour karyotype discriminates between good and bad prognostic outcome in neuroblastoma. Br J Cancer 1988; 57: 121-6.
Maris JM, White PS, Beltinger CP, Sulman EP, Castleberry RP, Shuster JJ, Look AT, Brodeur GM. Significance of chromosome 1p loss of heterozygosity in neuroblastoma. Cancer Res 1995; 55: 4664-9.
Caron H, van Sluis P, de Kraker J, Bokkerink J, Egeler M, Laureys G, Slater R, Westerveld A, Voute P.A, Versteeg R. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N Engl J Med 1996; 334: 225-30.
Plantaz D, Mohapatra G, Matthay KK, Pellarin M, Seeger RC, Feuerstein BG. Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization. Am J Pathol 1997; 150: 81-9.
Brinkschmidt C, Christiansen H, Terpe HJ, Simon R, Boecker W, Lampert F, Stoerkel S. Comparative genomic hybridization (CGH) analysis of neuroblastomas -an important methodological approach in paediatric tumour pathology. J Pathol 1997; 181: 394-400.
Vandesompele J, Van Roy N, Van Gele M, Laureys G, Ambros P, Heimann P, Devalck C, Schuuring E, Brock P, Otten J, Gyselinck J, De Paepe A, Speleman F. Genetic heterogeneity of neuroblastoma studied by comparative genomic hybridization. Genes Chromosomes Cancer 1998; 23: 141-52.
Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D, Meddeb M, Danglot G, Brinkschmidt C, Christiansen H, Laureys G, Speleman F, Nicholson J, Bernheim A, Betts DR, Vandesompele J, Van Roy N. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med 1999; 340: 1954-61.
Janoueix-Lerosey I, Penther D, Thioux M, de Cremoux P, Derre J, Ambros P, Vielh P, Benard J, Aurias A, Delattre O. Molecular analysis of chromosome arm 17q gain in neuroblastoma. Genes Chromosomes Cancer 2000; 28: 276-84.
Cunsolo CL, Bicocchi MP, Petti AR, Tonini GP. Numerical and structural aberrations in advanced neuroblastoma tumours by CGH analysis; survival correlates with chromosome 17 status. Br J Cancer 2000; 83: 1295-300.
Vandesompele J, Baudis M, De Preter K, Van Roy N, Ambros P, Bown N, Brinkschmidt C, Christiansen H, Combaret V, Lastowska M, Nicholson J, O’Meara A, Plantaz D, Stallings R, Brichard B, Van den Broecke C, De Bie S, De Paepe A, Laureys G, Speleman F. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J Clin Oncol 2005; 23: 2280-99.
Takita J, Hayashi Y, Kohno T, Shiseki M, Yamaguchi N, Hanada R, Yamamoto K, Yokota J. Allelotype of neuroblastoma. Oncogene 1995; 11: 1829-34.
Ejeskar K, Aburatani H, Abrahamsson J, Kogner P, Martinsson T. Loss of heterozygosity of 3p markers in neuroblastoma tumours implicate a tumour-suppressor locus distal to the FHIT gene. Br J Cancer 1998; 77: 1787-91.
Guo C, White PS, Weiss MJ, Hogarty MD, Thompson PM, Stram DO, Gerbing R, Matthay KK, Seeger RC, Brodeur GM, Maris JM. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999; 18: 4948-57.
Luttikhuis ME, Powell JE, Rees SA, Genus T, Chughtai S, Ramani P, Mann JR, McConville CM. Neuroblastomas with chromosome 11q loss and single copy MYCN comprise a biologically distinct group of tumours with adverse prognosis. Br J Cancer 2001; 85: 531-7.
Westermann F, Schwab M. Genetic parameters of neuroblastomas. Cancer Lett 2002; 184: 127-47.
Maris JM, Weiss MJ, Guo C, Gerbing RB, Stram DO, White PS, Hogarty MD, Sulman EP, Thompson PM, Lukens JN, Matthay KK, Seeger RC, Brodeur GM. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a Children’s Cancer Group study. J Clin Oncol 2000; 18: 1888-99.
Schwab M, Westermann F, Hero B, Berthold F. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol 2003; 4: 472-80.
Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003; 3: 203-16.
Castel V, Canete A, Navarro S, García-Miguel P, Melero C, Acha T, Navajas A, Badal MD. Outcome of high-risk neuroblastoma using a dose intensity approach: improvement in initial but not in long-term results. Med Pediatr Oncol 2001; 37: 537-42.
Kaneko M, Tsuchida Y, Mugishima H, Ohnuma N, Yamamoto K, Kawa K, Iwafuchi M, Sawada T, Suita S. Intensified chemotherapy increases the survival rates in patients with stage 4 neuroblastoma with MYCN amplification. J Pediatr Hematol Oncol 2002; 24: 613-21.
Castel V, Canete A, Noguera R, Navarro S, Oltra S. (Neuroblastoma). Clin Transl Oncol 2005; 7: 133-45.
Bocking A, Giroud F, Reith A. Consensus report of the European Society for Analytical Cellular Pathology task force on standardization of diagnostic DNA image cytometry. Anal Quant Cytol Histol 1995; 17: 1-7.
Navarro S, Noguera R, Pellin A, Mejía C, Ruiz A, Llombart-Bosch A. Pleomorphic anaplastic neuroblastoma. Med Pediatr Oncol 2000; 35: 498-502.
Noguera R, Canete A, Pellin A, Ruiz A, Tasso M, Navarro S, Castel V, Llombart-Bosch A. MYCN gain and MYCN amplification in a stage 4S neuroblastoma. Cancer Genet Cytogenet 2003; 140: 157-61.
Kallioniemi OP, Kallioniemi A, Piper J, Isola J, Waldman FM, Gray JW, Pinkel D. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer 1994; 10: 231-43.
Navarro S, Amann G, Beiske K, Cullinane CJ, DÁmore ESG, Gambini C, Mosseri V, De Bernardi B, Michon J, Peuchmaur M. Prognostic value of International Neuroblastoma Pathology Classification in localized respectable peripheral neuroblastic tumors: A histopathologic study of localized neuroblastoma European study group 94.01 trial and protocol. J Clin Oncol 2006; 42: 695-699.
Burgues O, Navarro S, Noguera R, Pellin A, Ruiz A, Castel V, Llombart Bosch A. Prognostic value of the International Neuroblastoma Pathology Classification in neuroblastoma (Schwannian stroma-poor) and comparison with other prognostic factors: a study of 182 cases from the Spanish Neuroblastoma Registry. Virchows Arch 2006; 449: 410-420.
Kallioniemi A, Kallioniemi O.P, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992; 258: 818-21.
Lastowska M, Nacheva E, McGuckin A, Curtis A, Grace C, Pearson A, Bown N. Comparative genomic hybridization study of primary neuroblastoma tumors. United Kingdom Children’s Cancer Study Group. Genes Chromosomes Cancer 1997; 18: 162-9.
Breen CJ, O’Meara A, McDermott M, Mullarkey M, Stallings RL. Coordinate deletion of chromosome 3p and 11q in neuroblastoma detected by comparative genomic hybridization. Cancer Genet Cytogenet 2000; 120: 44-9.
Plantaz D, Vandesompele J, Van Roy N, Lastowska M, Bown N, Combaret V, Favrot MC, Delattre O, Michon J, Benard J, Hartmann O, Nicholson JC, Ross FM, Brinkschmidt C, Laureys G, Caron H, Matthay KK, Feuerstein BG, Speleman F. Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer 2001; 91: 680-6.
Lastowska M, Cullinane C, Variend S, Cotterill S, Bown N, O’Neill S, Mazzocco K, Roberts P, Nicholson J, Ellershaw C, Pearson AD, Jackson MS. Comprehensive genetic and histopathologic study reveals three types of neuroblastoma tumors. J Clin Oncol 2001; 19: 3080-90.
Schleiermacher G, Janoueix-Lerosey I, Combaret V, Derre J, Couturier J, Aurias A, Delattre O. Combined 24-color karyotyping and comparative genomic hybridization analysis indicates predominant rearrangements of early replicating chromosome regions in neuroblastoma. Cancer Genet Cytogenet 2003; 141: 32-42.
Guo C, White PS, Hogarty MD, Brodeur GM, Gerbing R, Stram DO, Maris JM. Deletion of 11q23 is a frequent event in the evolution of MYCN single-copy high-risk neuroblastomas. Med Pediatr Oncol 2000; 35: 544-6.
Spitz R, Hero B, Ernestus K, Berthold F. FISH analyses for alterations in chromosomes 1, 2, 3, and 11 define high-risk groups in neuroblastoma. Med Pediatr Oncol 2003; 41: 30-5.
McArdle L, McDermott M, Purcell R, Grehan D, O’Meara A, Breatnach F, Catchpoole D, Culhane AC, Jeffery I, Gallagher WM, Stallings R.L. Oligonucleotide microarray analysis of gene expression in neuroblastoma displaying loss of chromosome 11q. Carcinogenesis 2004; 25: 1599-609.
Scaruffi P, Valent A, Schramm A, Astrahantseff K, Eggert A, Tonini GP. Application of microarray-based technology to neuroblastoma. Cancer Lett 2005; 228: 13-20.
Gotoh T, Hosoi H, Iehara T, Kuwahara Y, Osone S, Tsuchilla K, Ohira M, Nakagawara A, Kuroda H, Sugimoto T. Prediction of MYCN amplification in Neuroblastoma using serum DNA and real-time quantitative polymerase chain reaction. J Clin Oncol 2005) 23: 5205-5210.
![]()