
Vol. 40, n.º 3, 2007
|
REVISTA
ESPAÑOLA DE
Vol. 40, n.º 3, 2007 |
|
Homenaje
al Dr. Francisco José Martínez Tello |
REVISIONES
José I. López
Departamento de Anatomía Patológica. Hospital de
Cruces. Universidad del País Vasco (EHU/UPV). Plaza de Cruces, s/n. 48903
Cruces/Barakaldo.
joseignacio.lopezfernandezdevillaverde@osakidetza.net
RESUMEN
La conjunción de datos morfológicos y de hallazgos genéticos ha supuesto una gran evolución en la concepción y en el diagnóstico de las neoplasias renales, y todo ello se ha plasmado en la nueva clasificación de la OMS de 2004. El conocimiento de las alteraciones genéticas asociadas a un determinado tipo histológico de cáncer abre la puerta a nuevas terapéuticas, lo cual en el riñón alcanza una importancia capital debido a la tradicional quimio y radiorresistencia de sus carcinomas. La estrecha relación entre determinadas alteraciones genéticas y tipos histológicos concretos de carcinoma renal afianza el papel de la hematoxilina-eosina como método diagnóstico y confirma la importancia del patólogo en el diagnóstico de este grupo de tumores. Así, por mencionar sólo a los dos tipos histológicos mayoritarios y, expuestas de manera muy general, el carcinoma renal de células claras se asocia a diversas alteraciones en el cromosoma 3 y el carcinoma renal papilar a alteraciones en los cromosomas 7 y 17. Sin embargo, el análisis de estos dos tipos de cáncer renal desde el punto de vista genético ha complicado en parte el panorama diagnóstico con el reconocimiento por un lado de neoplasias intermedias, o híbridas, y por otro de discordancias graves entre la morfología y la genética. En este trabajo se revisa parte del estado actual del problema centrándolo en tres situaciones concretas, a saber, carcinomas renales de células claras que parecen papilares, carcinomas renales papilares que parecen de células claras, y verdaderos carcinomas mixtos papilares y de células claras, también llamados carcinomas renales híbridos.
Palabras clave: Carcinoma renal de células claras, carcinoma renal papilar, carcinoma renal híbrido, morfología, genética.
SUMMARY
The sum of morphological and genetic findings in renal cell neoplasia has led to a new evolution in their conception and diagnosis. This progress has been reflected in the last 2004 WHO classification of renal tumours. The recognition of genetic alterations associated to specific histological types of cancer opens the door for new therapies that in renal neoplasms are of paramount importance due to their well known chemo and radioresistance. The close relationship between some genetic alterations and specific histological types of renal cancer strengthens the role of the haematoxylin-eosin staining as a diagnostic method and confirms the pathologist’s importance in the diagnosis of this group of tumours. So, facing only with the two more common histological subtypes, clear cell renal cell carcinoma is associated to chromosome 3 alterations and papillary renal cell carcinomas does to chromosomes 7 and 17. However, the genetic analysis of these neoplasms in clinical practice has made their diagnosis more complicated because hybrid tumours and deep discordances between histological and genetic findings have appeared. This review analyses part of the state of the art in this topic by assessing three different approaches, i.e., clear cell renal cell carcinomas mimicking papillary renal cell carcinomas, papillary renal cell carcinomas remembering clear cell carcinomas, and true mixed, papillary and clear cell, renal cell carcinomas, also termed hybrid carcinomas.
Key words: Clear cell renal cell carcinoma, papillary renal cell carcinoma, hybrid renal carcinoma, histopathology, genetics.
INTRODUCCIÓN
El cáncer renal está situado entre los 10 cánceres más frecuentes en el varón (1) y todos los indicios apuntan a que su incidencia está creciendo en los últimos años (2). Desde el punto de vista clínico el cáncer renal se caracteriza por permanecer silente, u oligosintomático, durante largos períodos de tiempo, razón por la cual un número significativo de casos aparecen en estadios avanzados. Además es una neoplasia tradicionalmente resistente a tratamientos quimio o radioterápicos. Desde el punto de vista anatomopatológico el cáncer renal es extraordinariamente variopinto, con histologías muy diversas y no siempre bien clasificables, lo cual justifica tanto la rápida evolución de su clasificación morfológica desde los primeros intentos llevados a cabo por Thoenes en 1986 (3), como la importancia relativa del grupo de carcinomas inclasificables, que oscilaba en tiempos no lejanos entre el 10 y el 15% de los casos (3).
El advenimiento de los análisis genéticos y moleculares a la práctica diaria del patólogo ha supuesto una auténtica revolución en el campo de las neoplasias renales. De la conjunción de estos hallazgos con la histopatología clásica nace en 2004 la clasificación de los tumores renales de la OMS (4), la cual supone verdaderamente un punto y aparte en la concepción de la tumorigénesis renal. Esta nueva aproximación al problema clarifica algunas cosas, complica otras, y profundiza en todas, y este trabajo pretende ser un claro ejemplo de todo esto.
La detección de alteraciones genéticas específicas en cada tipo histológico de carcinoma renal supone una clarificación del problema. Así, las alteraciones localizadas en el cromosoma 3 se relacionan con el carcinoma renal de células claras (5), y las localizadas en los cromosomas 7 y 17 caracterizan al carcinoma renal papilar (6). Sin embargo, cuando la morfología no acompaña a los análisis genéticos, cuando los contradice, o cuando un tumor fenotípicamente bien encuadrado en una determinada categoría desde el punto de vista molecular manifiesta histologías mixtas, de células claras y papilar, por ejemplo, el problema se complica y un nuevo horizonte de incertidumbre diagnóstica aparece ante el patólogo. Esta revisión trata de incidir en este aspecto novedoso.
CARCINOMA RENAL DE CÉLULAS CLARAS CON ARQUITECTURA PAPILAR
Aproximadamente el 70 a 75% de los cánceres renales son carcinomas de células claras esporádicos (4). Es una neoplasia que afecta más frecuentemente a varones de raza negra y en general por encima de los 40 años de edad. El hábito tabáquico y la obesidad, esta última en mujeres, son factores predisponentes. Se postula que se originan a partir de la célula epitelial de ducto contorneado proximal (7).
Genética
Más del 90% de los carcinomas renales de células claras muestran la típica alteración citogenética en el brazo corto del cromosoma 3 (5), pudiendo encontrarse desde pérdidas completas del brazo hasta translocaciones o delecciones (8). Se ha demostrado que entre el 30% y el 56% de los carcinomas renales de células claras esporádicos tienen mutaciones en el gen de VHL y hasta el 19% tienen silenciamiento de mismo (9-11).
VHL es un gen supresor. Codifica una proteína con dos isoformas principales, pVHL30 y pVHL19, que son las responsables de la acción antitumoral del gen a través de la inactivación de los factores inductores de hipoxia en el citoplasma. Sin embargo, pVHL también tiene un papel en la regulación de las metaloproteinasas y de sus inhibidores, y de otras enzimas. Por ejemplo, la ausencia de pVHL incrementa los niveles de las anhidrasas carbónicas 9 y 12, sustancias que incrementan el nivel de acidificación del microambiente, lo cual favorece a su vez las propiedades invasoras y el crecimiento tumoral (12), y sobre-regulan la ciclina D1 (13). La presencia de anhidrasa carbónica 9 (CA9) y de ciclina D1 puede se ser determinada por métodos inmunohistoquímicos hoy en día en cortes de parafina, lo cual es importante para el patólogo en la practica diaria porque son enzimas cuya disregulación depende exclusivamente de alteraciones en el cromosoma 3.
Macroscopía
Los carcinomas renales de células claras presentan una coloración amarillenta y un aspecto multilobulado, con áreas sólidas y quísticas, y con hemorragia y necrosis, circunstancias que normalmente acompañan a los casos de gran tamaño. En estos casos, la pieza quirúrgica remitida puede llegar a pesar varios kilos. Sin embargo, esta circunstancia es hoy en día excepcional. La mayor concienciación de la población frente a los síntomas, la estructura sanitaria y el elevado grado de refinamiento que han alcanzado las técnicas de imagen, hacen que cada vez se detecten más casos confinados al riñón y de pequeño tamaño. Esto ha hecho posible el desarrollo en los últimos tiempos técnicas de crioablación o de ablación por radiofrecuencia por vía laparoscópica que minimizan los efectos de la cirugía convencional y que obtienen buenos resultados (14,15).
Los tumores suelen estar bien delimitados del tejido renal adyacente por una cápsula fibrosa que puede ser continua o no. Las áreas blanquecinas suelen estar relacionadas con la transformación sarcomatoide que ocurre en ocasiones en este tumor. Las áreas de coloración parda suelen corresponder a zonas de hemorragia antigua. Alrededor del 15% de los casos tienen calcificaciones. La presencia de hueso metaplásico es muy poco frecuente.
Ocasionalmente, el carcinoma de células claras está constituido por quistes hasta el punto de ser confundido con lesiones no tumorales. Se trata de la variante multilocular, un subtipo de carcinoma de células claras muy poco frecuente y de bajo grado.
El carcinoma renal de células claras asociado a la enfermedad de von Hippel-Lindau suele ser con frecuencia multifocal, bilateral y quístico, y aparece en edades más tempranas, por lo cual habrá que pensar en esta enfermedad cuando nos encontremos con tumores renales con estas características en personas más jóvenes de lo esperado (16).
Histopatología
El carcinoma de células claras muestra una histología altamente característica. Las células del carcinoma de células claras muestran un citoplasma óptimamente vacío y una membrana citoplásmica muy bien definida. En ocasiones las células muestran citoplasmas débilmente eosinófilos en torno al núcleo, circunstancia observada en casos de alto grado nuclear. Si este cambio eosinofílico del citoplasma es muy extenso, pueden plantearse dudas diagnósticas iniciales con el carcinoma renal de células cromófobas. El núcleo puede mostrar morfologías variables y dependiendo de su morfología, de sus irregularidades y de la presencia de nucleolo, se ha establecido el sistema de Furhman (17) para determinar el grado. Esta forma de graduar los carcinomas renales está ampliamente extendida entre los patólogos de todo el mundo con resultados altamente reproducibles, aunque existen intentos recientes para modificar en parte esta forma de asignar el grado histológico a estos tumores (18,19).
Las células se disponen formando nidos, estructuras alveolares y cordones o tapizando quistes de tamaño variable. El estroma contiene una red capilar muy prominente y típica. Las zonas centrales del tumor pueden mostrar áreas cicatriciales, esclerosis o edema. Es frecuente la hemorragia y la necrosis, sobre todo en los tumores grandes. Es típico encontrar acúmulos de hematíes en las luces de las estructuras alveolares. La transformación sarcomatoide puede producirse de forma brusca, aunque lo normal es que se puedan encontrar áreas de transición si se muestrea el tumor suficientemente. Estas áreas pueden presentar morfologías variadas, pero predomina el fenotipo fusocelular.
La variedad multilocular del carcinoma renal de células claras exige para su diagnóstico la ausencia total de áreas sólidas de tumor, estando este enteramente constituido por finas paredes tapizadas por células claras típicas, normalmente de bajo grado nuclear. La presencia, aunque sea minoritaria, de zonas sólidas de tumor debe llevar al patólogo al diagnóstico de carcinoma renal de células claras convencional, sin más apellidos.
El problema en la interpretación diagnóstica de esta variedad histológica puede aparecer cuando el aspecto histológico no sea típico, por ejemplo, cuando el tumor muestre áreas de arquitectura preferentemente papilar, citoplasmas no del todo «aclarados», o cuando al lado de un carcinoma de células claras convencional se observen zonas de aspecto diferente, con papilas, con túbulos, o con células eosinófilas que no cuadran con lo esperado en un carcinoma de células claras (fig. 1). En estos casos el patólogo puede dudar y diagnosticar bien un carcinoma de células claras (fig. 2), bien un carcinoma papilar (fig. 3), o bien un carcinoma inclasificable atendiendo a que estos últimos también incluyen a aquellos casos complejos que muestren dos o más histologías. La interpretación diagnóstica del patólogo en estos casos es totalmente subjetiva, y en ocasiones depende simplemente de las cantidades relativas de cada fenotipo, lo cual, no lo olvidemos, depende a su vez del azar en el muestreo en la sala de macros. Hay que señalar que volver a la sala de macros para incluir más tejido no suele solucionar el dilema de qué nombre concreto poner a estos casos, vamos a decir, «complicados».
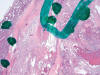
Fig. 1:
Imagen panorámica de un carcinoma renal de células claras con áreas típicas de
célula clara (círculo) y áreas papilares (puntos) (Hematoxilina-eosina, x25).
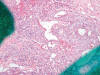
Fig. 2:
Detalle histológico de un carcinoma de células claras mostrando un área de
células claras típica (correspondiente a la zona del círculo de la figura 1)
(Hematoxilina-eosina, x250).
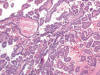
Fig. 3:
Detalle histológico de un carcinoma de células claras mostrando un área papilar
(correspondiente a la zona de puntos de la figura 1) (Hematoxilina-eosina,
x250).
La primera mención específica a un carcinoma de este tipo aparece en la literatura en 1999 (20), en la que los autores detallan 3 casos de carcinoma renal papilar con células claras, pérdidas de segmentos cromosómicos en 3p, monosomía 14 y ausencia de trisomías 7 y 17. Los autores concluyen que, a pesar de la arquitectura papilar predominante en los casos estudiados, los hallazgos citogenéticos demuestran que se trata de un carcinoma de células claras. Posteriormente, Salama y cols en 2003 (21) llegan a la misma conclusión en un estudio de 7 tumores en 6 pacientes. Aunque poco frecuente, la posibilidad de encontrar un caso de este tipo es real en nuestro medio. De hecho, en la serie de Mai y cols (22) esta peculiaridad histológica se produce en aproximadamente el 2,7% de los carcinomas renales de células claras.
Patología molecular e inmunohistoquímica
La solución pasa por realizar un estudio molecular del tumor, seleccionando para ello tanto las áreas típicas de células claras como las zonas problemáticas, las zonas papilares, las células de citoplasma no claro, etc., y demostrar que, independientemente de la morfología, todas ellas presentan alteraciones en el cromosoma 3. Esto certifica que el caso problema es todo él un carcinoma de células claras, eso sí, con áreas de arquitectura papilar. Sin embargo, y por razones obvias, esta solución aún está al alcance de pocos laboratorios en nuestro entorno y puede resultar poco factible en la dinámica rutinaria de la mayoría de nuestros hospitales.
La inmunohistoquímica es mucho más asequible y también ayuda porque además de coexpresar citoqueratinas de bajo peso molecular (CK 8, 18, 19) y vimentina, y de marcar con EMA, con el marcador de carcinoma renal (RCCm), con CD10 y CD13, con CD15 y con MUC-1 (23), la tinción con CA9 (figs. 4 y 5) y con ciclina D1 (figs. 6 y 7), marcadores dependientes exclusivamente de la alteración en el cromosoma 3, también es positiva. Por último, resultan negativas las inmunotinciones con CK7, CK14, 34betaE12, CK20, parvalbúmina, CD117, racemasa (p504S) y la e-cadherina (23).

Fig. 4:
Carcinoma de células claras mostrando positividad de membrana con anhidrasa
carbónica 9 (CA9) en la zona de células claras (correspondiente a la zona del
círculo de la figura 1) (CA9, x250).

Fig. 5:
Carcinoma de células claras mostrando positividad de membrana con anhidrasa
carbónica 9 (CA9) en la zona papilar (correspondiente a la zona de puntos de la
figura 1) (CA9, x250).
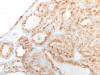
Fig. 6:
Carcinoma de células claras mostrando positividad nuclear con ciclina D1 en la
zona de células claras (correspondiente a la zona del círculo de la figura 1)
(ciclina D1, x250).
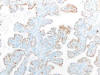
Fig. 7:
Carcinoma de células claras mostrando positividad nuclear con ciclina D1 en la
zona papilar (correspondiente a la zona de puntos de la figura 1) (ciclina D1,
x250).
CARCINOMA RENAL PAPILAR CON CÉLULAS CLARAS
Aproximadamente el 10 a 15% de los carcinomas renales primarios son carcinomas papilares (4). En comparación con el carcinoma renal de células claras, el carcinoma renal papilar tiene mejor comportamiento biológico (24). La primera descripción detallada de estos tumores apareció en 1976 (25). Con posterioridad, la clasificación morfológica de los tumores renales propuesta por Thoenes y cols (3) los denominó carcinomas renales cromófilos debido a la basofilia citoplásmica que exhibían. La clasificación de Mainz (26) supuso una evolución integrando los primeros hallazgos citogenéticos. El primer intento de separar los carcinomas renales papilares en dos subgrupos en función de datos exclusivamente morfológicos corrió a cargo de Delahunt y Eble (27). Muy poco después dicha subclasificación histológica se reforzó con el hallazgo de alteraciones genéticas específicas de cada uno de los dos tipos previamente descritos (6).
Genética
El carcinoma renal papilar está caracterizado por presentar trisomías o tetrasomías de los cromosomas 7 y 17 y por pérdida del cromosoma Y (26, 28). Asimismo, se detectan ganancias cromosómicas de 3q, 12, 16 y 20 (28, 29). La trisomía 7 es característica pero no específica y se puede encontrar también en tumores de vejiga, próstata y de la cabeza y cuello, además de en la hiperplasia adenomiomatosa de próstata y en otras enfermedades benignas. Sin embargo es una alteración muy constante tanto en las fases tempranas como tardías de la enfermedad, lo que sugiere que los genes situados en el cromosoma 7 juegan un papel fundamental en la patogenia de estos tumores (30). La trisomía 17 es mucho más específica y valorable desde el punto de vista diagnóstico, pudiendo encontrarse trisomías completas, isocromosomas 17q, o duplicaciones en 17q21.
El análisis genético del carcinoma renal papilar sirve además como elemento predictivo de pronóstico y para ahondar en el conocimiento de la neoplasia. Por ejemplo, los carcinomas papilares con pérdida de Xp (6) o de 9p (31) tienen mal pronóstico, con supervivencias acortadas respecto del resto. Gunawan y cols (31) han demostrado mediante estudios citogenéticos que el tipo 2 de carcinoma renal papilar surge de una evolución a partir del tipo 1, y Jones y cols. (32) encuentran que, a diferencia de los carcinomas de células claras multifocales que se consideran de origen único y en los que la multifocalidad se explicaría como un proceso metastático intrarrenal, los carcinomas papilares multifocales tienen un origen independiente.
Macroscopía
Se trata de neoplasias bien circunscritas y generalmente excéntricas en el riñón. La mayoría están confinadas al riñón y son encapsuladas (33). Al corte son tumores de coloración heterogénea dependiendo del grado de necrosis, hemorragia, y de la cantidad de histiocitos espumosos acumulada, variando desde el color amarillento hasta en marrón o rojizo. Es típico encontrar necrosis y hemorragia extensas, aunque este hecho no parece conferirle un peor pronóstico (34). La necrosis puede ser tan extensa que la imagen radiológica puede ser enteramente quística y los radiólogos a veces encuentran dificultades para etiquetar la masa como un tumor. El examen macroscópico en estos casos con necrosis extensa obliga a una minuciosa búsqueda de zonas conservadas, generalmente en la periferia, para encontrar tejido viable y poder diagnosticar mejor. Los carcinomas papilares se asocian con mayor frecuencia que los demás tumores renales a adenomas corticales. La multifocalidad, encontrada con cierta frecuencia en este tipo de tumor tampoco parece conferir peor pronóstico, y su presencia no debe ser un argumento contra la cirugía conservadora (35). Existen casos aislados que asocian carcinomas y adenomas papilares múltiples en los que algunos tumores muestran histologías mixtas, a caballo entre adenoma y carcinoma, sugiriendo una transición entre ambos (36).
Histopatología
El carcinoma renal papilar tipo 1 es el más frecuente. Está constituido por células pequeñas, con escaso citoplasma basófilo y núcleo redondeado sin nucleolo, que se aponen en hilera simple en el eje hialino-vascular de la papila. Se trata de tumores de grado bajo que contienen abundantes acúmulos de histiocitos espumosos y cuerpos de psammoma en las papilas.
El carcinoma renal papilar tipo 2 se caracteriza por papilas irregulares y células de gran tamaño con citoplasma amplio y eosinofílico y núcleo voluminoso con nucleolo prominente. Las células a menudo muestran imágenes de pseudoestratificación en las papilas. Los cuerpos de psammoma y los acúmulos histiocitarios aparecen en menor cantidad que en el tipo 1.
Normalmente el grado (17) es mayor en el tipo 2 que en el tipo 1 (37). Recientemente se ha propuesto que los carcinomas renales papilares deben ser graduados en base al grado nucleolar y no mediante el sistema de Furhman (38). Asimismo, el estadio TNM (39) suele ser más avanzado en el tipo 2 (37).
Como el resto de carcinomas renales, el carcinoma papilar puede presentar transformación sarcomatoide (40). En ocasiones el tumor es de morfología sólida (41) o mixta, tubular y papilar y, más raramente, de predominio tubular y en este terreno pueden producirse ciertas dificultades diagnósticas. A veces muestran transformación oncocítica de sus células (42). Existen controversias en la literatura reciente sobre si considerar o no al carcinoma fusocelular tubular y mucinoso, un tipo específico de carcinoma renal dentro de la última clasificación de la OMS (4), como un subtipo de carcinoma papilar o no. La literatura presenta opiniones encontradas y probablemente hagan falta más casos y más estudios para dilucidar el problema. Por ejemplo, mientras Cossu-Rocca y cols. (43) descartan esta posibilidad basándose en estudios genéticos, Shen y cols. (44) la apoyan.
La aparición focal de células claras en el carcinoma papilar es un hecho relativamente frecuente que no importuna para el diagnóstico, pero cuando el cambio es abundante comienzan los problemas interpretativos: ¿se trata de un carcinoma papilar?, ¿es un carcinoma de células claras?, ¿y por qué no un carcinoma inclasificable? (fig. 8). La histología en la hematoxilina-eosina y el dilema diagnóstico son los mismos que los detallados en el grupo anterior de tumores, pero la realidad que subyace es exactamente la inversa, es decir son verdaderos tumores papilares con áreas de células claras. En la serie de Mai y cols. (22), este tipo de situación supone cerca del 5% de los casos.
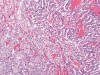
Fig. 8:
Imagen representativa de un carcinoma renal papilar con áreas papilares y áreas
de células claras (hematoxilina-eosina x100).
Patología molecular e inmunohistoquímica
El estudio citogenético de todas las áreas de un carcinoma renal papilar genuino mostrará los cambios característicos de esta variedad, sobre todo trisomías del 7 y 17, y pérdida del Y, que son las más constantes, sin alteraciones en el cromosoma 3.
El carcinoma renal papilar comparte con el carcinoma renal de células claras la mayor parte de los marcadores inmunohistoquímicos al uso. Solo la expresión de CK7 (fig. 9) y de racemasa (p504S) los distingue con fiabilidad, siendo positivas en el primero y negativa en el segundo. Tanto el tipo 1 como el tipo 2 resultan positivos con CD10, con el marcador de carcinoma renal (RCCm), con citoqueratinas de bajo peso y con p504S, y muestran inmunotinción negativa para CK20 y citoqueratinas de alto peso molecular. El tipo 1, además, es positivo con CK7 y MUC1 (45), y el tipo 2 lo es con e-cadherina (23). Tanto CA9 como la ciclina D1 son siempre negativos.
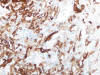
Fig. 9:
Carcinoma renal papilar mostrando Intensa positividad con CK7 en las áreas de
células claras (CK7, x250).
CARCINOMA RENAL HÍBRIDO, PAPILAR Y DE CÉLULAS CLARAS
Los tumores híbridos, es decir, aquellos tumores únicos que en su constitución intervienen dos o más variedades histológicas íntimamente entremezcladas, están de moda en patología. No se trata de tumores dobles, ni de tumores de colisión, y afectan a múltiples órganos y tejidos, siendo probablemente el sistema endocrino el más representativo de todos.
En el riñón prácticamente todas las posibilidades combinatorias están abiertas, pero quizás el ejemplo más claro de esta situación está representado por la dualidad entre el carcinoma de células cromófobas y el oncocitoma en el contexto del síndrome de Birt-Hogg-Dubé (46), por ejemplo, y por toda la gama de situaciones intermedias que intentan demostrar que ambos tumores son los miembros extremos de un espectro único de neoplasias originadas en la nefrona distal (47-52). También se han documentado casos de tumores híbridos compuestos por carcinoma papilar y tumor oncocítico (42,53-55) y, más ocasionalmente, híbridos de variedades propias de la nefrona proximal, es decir, mezclas de carcinoma papilar y de carcinoma de células claras (22).
En estos casos, ambos tipos histológicos están perfectamente representados dentro del mismo tumor (fig. 10), con áreas papilares generalmente de tipo 1 (fig. 11) conteniendo acúmulos de histiocitos espumosos y áreas de células claras (fig. 12), con arquitectura en nidos y patrón vascular característico. Las áreas papilares muestran trisomías 7 y 17, pérdida del cromosoma Y, e inmunopositividad para CK7 (fig. 13) y racemasa, y las áreas de células claras pérdida de 3p u otras alteraciones en este cromosoma, e inmunorreactividad con CA9 (fig. 14) y ciclina D1.
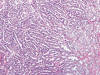
Fig. 10:
Imagen representativa de un carcinoma renal híbrido (Hematoxilina-eosina, x100).
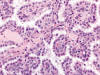
Fig. 11:
Detalle histológico del área papilar del carcinoma renal híbrido (zona izquierda
en la figura 12) (Hematoxilina-eosina, x250).
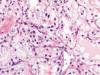
Fig. 12:
Detalle histológico del área de células claras del carcinoma renal híbrido (zona
derecha de la figura 12) (Hematoxilina-eosina, x250).

Fig. 13:
Carcinoma renal híbrido mostrando intensa positividad para CK7 en la zona
papilar (CK7, x250).

Fig. 14:
Carcinoma renal híbrido mostrando positividad de membrana para anhidrasa
carbónica 9 en la zona de células claras (CA9, x250).
En la serie de Mai y cols. (22), el 4,8% de los carcinomas renales tienen áreas de células claras y áreas papilares y, en su opinión este grupo de tumores tiene un comportamiento biológico más agresivo.
Existen, asimismo, tumores híbridos compuestos por carcinoma mucinoso, tubular y fusocelular y carcinoma de células claras (56), y por carcinoma papilar asociado a leiomiomatosis hereditaria y carcinoma de células claras (57).
CONCUSIONES
En este trabajo se han revisado las posibilidades diagnósticas que se esconden detrás de una misma constelación histológica en el cáncer renal. Así, un contexto morfológico que incluya células claras y papilas en cantidad y disposición variable puede esconder igualmente un carcinoma de células claras con patrón de crecimiento papilar, un carcinoma papilar que contenga células claras, o un verdadero carcinoma renal híbrido, con áreas genuinas de cada una de las dos variedades anteriores. Se trata de una situación con la que el patólogo se enfrenta con cierta frecuencia y a la que a menudo se da soluciones poco o nada estandarizadas, basadas más en el olfato momentáneo que en criterios morfológicos. Hoy en día, la trascendencia clínica de todo esto es prácticamente nula debido a que los pacientes afectos de cáncer renal entran todos ellos en la misma dinámica de seguimiento por parte de urólogos y oncólogos, pero en el futuro puede tener importancia vista la rápida evolución de los tratamientos más modernos.
La llegada de la patología molecular a nuestra especialidad y su implicación en la nueva clasificación del cáncer renal de la OMS de 2004 coloca a los patólogos ante un nuevo panorama diagnóstico. El conocimiento previo de estas posibles variaciones puede ayudar a interpretar mejor resultados inmunohistoquímicos incompletos, inesperados o mixtos. El análisis de las diversas zonas de cada tumor incluyendo CA9, ciclina D1, racemasa y CK7 puede resultar una ayuda extra, aunque el análisis molecular de las áreas problemáticas dilucidará finalmente el diagnóstico.
BIBLIOGRAFÍA
Jemal A, Siegel R, Ward E, y cols. Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43-66.
Chow WH, Devesa SS, Warren JL, y cols. Rising incidenceof renal cell cancer in the United States. JAMA 1999; 281: 1628-31.
Thoenes W, Störkel S, Rumpelt HJ. Histopathology and classification of renal cell tumors (adenomas, oncocytomas, and carcinomas). Path Res Pract 1986; 181: 125-43.
López-Beltrán A, Scarpelli M, Montironi R, y cols. 2004 WHO classification of the renal tumors of the adults. Eur Urol 2006; 49: 798-805.
Anglard P, Troy K, Brauch H, y cols Molecular analysis of genetic changes in the origin and development of renal cell carcinoma. Cancer Res 1991; 51: 1071-7.
Jiang F, Richter J, Schraml P, y cols. Chromosomal imbalances in papillary renal cell carcinoma: Genetic differences between histologic types. Am J Pathol 1998; 153: 1467-73.
Yoshida SO, Imam A, Olson CA y cols. Proximal renal tubular surface membrane antigens identified in primary and metastatic renal cell carcinomas. Arch Pathol Lab Med 1986; 110: 825-32.
Yoshida MA, Ohyashiki K, Ochi H y cols. Cytogenetic studies of tumor tissue from patients with nonfamilial renal cell carcinoma. Cancer Res 1986; 46: 2139-47.
Foster K, Prowse A, van der Berg A, y cols. Somatic mutations of the von Hippel-Lindau disease tumour suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet 1994; 3: 2169-73.
Schraml P, Struckmann K, Hatz F, y cols. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J Pathol 2002; 196: 186-93.
Bottaro DP, Linehan WM. Multifocal renal cancer: genetic basis and its medical relevance. Clin Cancer Res 2005; 11: 7206-8.
Lam JS, Shvarts O, Said JW y cols Clinicopathologic and molecular correlations of necrosis in the primary tumor of patients with renal cell carcinoma. Cancer 2005; 103: 2517-25.
Sufan RI, Jewett MAS, Ohh M. The role of von Hippel-Lindau tumor suppressor protein and hypoxia in renal clear cell carcinoma. Am J Physiol Renal Physiol 2004; 287: 1-6.
Park S, Cadeddu JA, Shingleton WB. Oncologic outcomes for ablative therapy of kidney cancer. Curr Urol Rep 2007; 8: 31-7.
López JI, Fernández de Larrinoa A, Zabala R, Oleada L, del Cura JL, Bilbao FJ. Utilidad de la biopsia cilindro guiada por control ecográfico en el diagnóstico y manejo de los tumores renales. Rev Esp Patol 2006; 39: 219-27.
Grignon DJ, Che M. Clear cell renal cell carcinoma. Clin Lab Med 2005; 25: 305-16.
Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 1982; 6: 655-63.
Novara G, Martignoni G, Artibani W, Ficarra V. Grading systems in renal cell carcinoma. J Urol 2007; 177: 430-6.
Rioux-Leclercq N, Karakiewicz PI, Trinh QD, y cols. Prognostic ability of simplified nuclear grading on renal cell carcinoma. Cancer 2007; 109: 868-74.
Füzesi L, Gunawan B, Bergmann F, Tack S, Braun TS, Jakse G. Papillary renal cell carcinoma with clear cell cytomorphology and chromosomal loss of 3p. Histopathology 1999; 35: 157-61.
Salama ME, Worsham MJ, DePeralta-Venturina M. Malignant papillary renal tumors with extensive clear cell change. A molecular analysis by microsatelite analysis and fluorescence in situ hybridization. Arch Pathol Lab Med 2003; 127: 1176-81.
Mai KT, Kohler DM, Roustan Delatour NL, Veinot JP. Cytohistopathologic hybrid renal cell carcinoma with papillary and clear cell features. Pathol Res Pract 2006; 202: 863-8.
Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical maarkers in the diferencial diagnosis of renal neoplasms. Clin Lab Med 2005; 25: 247-57.
Pignot G, Elie C, Conquy S, y cols. Survival analysis of 130 patients with papillary renal cell carcinoma: Prognostic utility of type 1 and type 2 subclassification. Urology 2007; 69: 230-5.
Mancilla-Jimenez R, Stanley RJ, Blath RA. Papillary renal cell carcinoma. A clinical, radiologic and pathologic study of 34 cases. Cancer 1976; 38: 2469-80.
Kovacs G. Molecular differential pathology of renal cell tumours. Histopathology 1993; 22: 1-8.
Delahunt B, Eble JN. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997; 10: 537-44.
Kovacs G, Füzesi L, Emanual A, y cols. Cytogenetics of papillary renal cell tumors. Genes Chromosomes Cancer 1991; 3: 249-55.
Henke RP, Erbersdobler A. Numerical chromosomal aberrations in papillary renal cortical tumors. Relationship with histopathologic features. Virchows Arch 2002; 440: 604-9.
Glukhova L, Lavialle C, Fauvet D, y cols. Mapping of the 7q31 subregion common to the small chromosome 7 derivatives from two sporadic papillary renal cell carcinomas: increased copy number and overexpression of the MET proto-oncogene. Oncogene 2000; 19: 754-61.
Gunawan B, von Heydebreck A, Fritsch T, y cols. Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of Type 2 from Type 1 tumors. Cancer Res 2003; 63: 6200-5.
Jones TD, Eble JN, Wang M, y cols. Molecular genetic evidence for the independent origin of multifocal papillary tumors in patients with papillary renal cell carcinomas. Clin Cancer Res 2005; 11: 7226-33.
Murphy WM, Grignon DJ, Perlman EJ, editores. Tumors of the Kidney, Bladder, and Related Urinary Structures. AFIP Atlas of Tumor Pathology, 4th Series, Fascicle 1. Washington DC: American Registry of Pathology; 2004.
Lee SE, Byun SS, Oh JK, y cols. Significance of macroscopic tumor necrosis as a prognostic indicador for renal cell carcinoma. J Urol 2006; 176: 1332-7.
Mejean A, Hopirtean V, Bazin JP, y cols. Prognostic factors for the survival of patients with papillary renal cell carcinoma: meaning of histological typing and multifocality. J Urol 2003; 170: 764-7.
Kiyoshima K, Oda Y, Nakamura T, y cols. Multicentric papillary renal cell carcinoma associated with renal adenomatosis. Pathol Int 2004; 54: 266-272.
Delahunt B, Eble JN, McCredie MRE, y cols. Morphologic typing of papillary renal cell carcinoma: Comparison of growth kinetics and patient survival in 66 cases. Hum Pathol 2001; 32: 590-5.
Sika-Paotonu D, Bethwaite PB, McCredie MRE, y cols. Nucleolar grade but not Furhman grade is applicable to papillary renal cell carcinoma. Am J Surg Pathol 2006; 30: 1091-6.
Sobin LH, Wittekind C, editores. TNM classification of Malignant Tumours, 6th edition, New Jersey: John Wiley & Sons; 2002.
Cohen RJ, McNeal JE, Susman M, y cols. Sarcomatoid renal cell carcinoma of papillary origin. A case report and cytogenetic evaluation. Arch Pathol Lab Med 2000; 124: 1830-2.
Renshaw AA, Zhang H, Corless CL, y cols. Solid variants of papillary (chromophil) renal cell carcinoma: clinicopathologic and genetic features. Am J Surg Pathol 1997; 21: 1203-9.
Lefevre M, Couturier J, Sibony M, Bazille C, Boyer K, Callard P, Vieillefond A, Allory Y. Adult papillary renal tumor with oncocytic cells. Clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol 2005; 29: 1576-81.
Cossu-Rocca P, Eble JN, Delahunt B, y cols. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006; 19: 488-93.
Shen SS, Ro JY, Tamboli P, y cols. Mucinous tubular and spindle cell carcinoma of kidney is probable a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol 2007; 11: 13-21.
Leroy X, Zini L, Leteurtre E, y cols. Morphologic subtyping of papillary renal cell carcinoma: correlation with prognosis and differential expresión of MUC1 between the two subtypes. Mod Pathol 2003; 15: 1126-30.
Adley BP, Schafernak KT, Yeldandi AV, Yang XJ, Nayar R. Cytologic and histologic findings in multiple renal hybrid oncocytic tumors in a patient with Birt-Hogg-Dubé syndrome: a case report. Acta Cytol 2006; 50: 584-88.
Abrahams NA, Tamboli P. Oncocytic renal neoplasms: diagnostic considerations. Clin Lab Med 2005; 25: 317-39.
Mai KT, Dhamanaskar P, Belanger E, Stinson WA. Hybrid chromophobe renal cell neoplasm. Pathol Res Pract 2005; 201: 385-9.
Salido M, Lloreta J, Melero C, y cols. Insertion (8;11) in a renal oncocytoma with multifocal transformation to chromophobe renal cell carcinoma. Cancer Genet Cytogenet 2005; 163: 160-3.
Nagashima Y, Mitsuya T, Shioi K, y cols. Renal Oncocytosis. Pathol Int 2005; 55: 210-5.
Al-Saleem T, Cairns P, Dulaimi AE, y cols The genetics of renal oncocytosis: a posible model for neoplastic progresión. Cancer Genet Cytogenet 2004; 152: 23-8.
Patton KT, Tretiakova MS, Yao JL, y cols. Expression of RON proto-oncogene in renal oncocytoma and chromophobe renal cell carcinoma. Am J Surg Pathol 2004; 28: 1045-50.
Al-Saleem T, Balsara BR, Liu Z, Feder M, Testa JR, Wu H, Greenberg RE. Renal oncocytoma with loss of chromosomes Y and 1 evolving to papillary carcinoma in connection with gain of chromosome 7. Coincidence or progression? Cancer Genet Cytogenet 2005; 163: 81-5.
Hes O, Brunelli M, Michal M, Cossu-Rocca P, Hora M, Chilosi M, Mina M, Boudova L, Menestrina F, Martignoni G. Oncocytic papillary renal cell carcinoma: a clinicopathologic, immunohistochemical, ultrastructural and interphase cytogenetic study of 12 cases. Ann Diagn Pathol 2006; 10: 133-9.
Rowsell C, Fleshner N, Marrano P, Squire J, Evans A. Papillary renal cell carcinoma within a renal oncocytoma: case report of an incidental finding of a tumour within a tumour. J Clin Pathol 2007; 60: 426-8.
Hes O, Hora M, Perez-Montiel DM, y cols. Spindle and cuboidal renal cell carcinoma, a tumor having frequent association with nephrolithiasis: report of 11 cases including a case with hybrid conventional renal cell carcinoma/spindle and cuboidal renal cell carcinoma components. Histopathology 2002; 41: 549-55.
Lehtonen HJ, Blanco I, Piulats JM, Herva R, Launonen V, Aaltonen LA. Convencional renal cell cancer in a patient with fumarate hydratase mutation. Hum Pathol 2007; 38: 793-6.
![]()