
Vol. 40, n.º 4, 2007
|
REVISTA
ESPAÑOLA DE
Vol. 40, n.º 4, 2007 |
CASUÍSTICA
Juan Carlos López Duque, Mónica Saiz Camín, Juan José Burgos Bretones, José M.ª Rivera-Pomar
Servicios de Anatomía Patológica del Hospital de Cruces.
Universidad del País Vasco. Plaza de Cruces, s/n. 48903 Baracaldo (Vizcaya).
juanc@jet.es
RESUMEN
El blastoma pleuropulmonar (BPP) es un tumor disontogénico pediátrico muy raro y agresivo reconocido como entidad clínico-patológica desde 1988 y del que se han descrito alrededor de 100 casos en la literatura mundial. Se clasifica como tipo I, II y III en base a su naturaleza quística o sólida. Presentamos un caso de una niña de 3 años con una tumoración que ocupa gran parte del hemitórax izquierdo con colapso del pulmón izquierdo y derrame pleural. Macroscópicamente es una masa sólida con áreas quísticas de 400 g y 15 cm de diámetro. Microscópicamente se trata de una tumoración mesenquimal de diferenciación divergente con zonas blastematosas y mesenquimales que incluye diferenciación rabdomioblástica, fibroblástica, liposarcomatosa y condroblástica-condroide. Su inmunofenotipo muestra positividad para desmina en extensas áreas del tumor, actina estriada en célula aisladas y S-100 en los focos condroides. Marcadores neuroendocrinos negativos. Queratina positiva únicamente en las células epiteliales y mesoteliales de revestimiento atrapadas en el crecimiento tumoral.
Palabras clave: Blastoma pleuropulmonar, tumores disontogénicos, pulmón.
SUMMARY
Pleuropulmonary blastoma is an aggressive and extremely rare pediatric dysontogenic tumor. It was firstly described in 1988 and since then only about 100 cases have been reported. Based on of cystic or solid predominant features, type I, type II and type III were established. In this paper a 3 yrs old female case is reported. The tumor mass occupied the main part of left hemithorax, collapsing lung and causing pleural effusion. Surgical specimen was a solid with cystic areas, 400 g and 15 cm in main diameter mass. Microscopically it was a mesenchymal tumor showing blastematous areas and divergent differentiation with rhabdomyoblastic, fibroblastic, liposarcomatous and chondroblastic/chondroid components. Immunohistochemistry showed positivity for desmin in wide areas, striated muscle actin in isolated cells and S-100 in chondroid foci. Neuroendocrine markers were negative. Cytokeratins were positive only in epithelial and mesothelial cells remaining within the tumor.
Key words: Pleuropulmonary blastoma, dysontogenic tumors, lung.
INTRODUCCIÓN
El blastoma pleuropulmonar (BPP) es un tumor intratorácico maligno muy poco frecuente que aparece, casi exclusivamente, en niños menores de 6 años (4). Descrito como entidad clínico-patológica en 1988 por Manivel (1), se trata de una neoplasia constituida por elementos mesenquimales primitivos embrionarios, incluyendo células indiferenciadas tipo blastema y áreas de diferenciación cartilaginosa, rabdomioblástica y/o fibroblástica. Se distinguen tres tipos de BPP según la configuración del tumor: I (quístico), II (mixto) y III (sólido). Se encuentra encuadrado dentro de las neoplasias disembriónicas (13).
DESCRIPCION DEL CASO
La paciente, una niña de 3 años es remitida a nuestro hospital con historia previa de cuadro febril de 10 días de evolución que persiste, a pesar de tratamiento antibiótico por sospecha de faringoamigdalitis. En la radiografía simple de tórax presenta una opacificación del hemitórax izquierdo con desviación del mediastino hacia el lado derecho (fig. 1-A). En la resonancia magnética se observa una tumoración que ocupa la gran mayoría del hemitórax izquierdo, sólida, en general homogénea presentando únicamente algunas pequeñas zonas de aspecto microquístico. La masa desplaza y colapsa al pulmón izquierdo y se acompaña de derrame pleural (fig. 1-B). Se programa a la paciente para intervención que cursa sin incidencias destacables.
![]()
Fig. 1:
Radiografía simple de tórax con borramiento del hemitórax izquierdo y
desplazamiento del mediastino hacia el lado derecho (A). En la resonancia
magnética se aprecia una masa redondeada, de límites precisos, sólida, con
algunas pequeñas zonas cavitadas que desplaza y comprime al pulmón hasta
colapsarlo casi por completo. Alrededor del tumor se observan signos de derrame
pleural (B).
El material quirúrgico remitido al servicio de Anatomía Patológica, está formado por varios fragmentos que en conjunto tienen un diámetro de 15 cm y un peso de 400 g, también se reciben muestras procedentes de diferentes zonas del diafragma. Externamente la masa principal presenta una superficie lisa y ligeramente lobulada. A la sección la superficie de corte es heterogénea con áreas blanquecinas de aspecto esponjoso y brillante, mucoide, con presencia ocasional de cavidades quísticas irregulares de pequeño tamaño. El tumor muestra amplias áreas hemorrágico-necróticas de coloración marrón rojiza y consistencia blanda y friable (fig. 2).
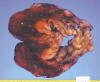
Fig. 2:
El tumor aunque fragmentado, tiene aspecto lobular de superficie lisa, a la
sección es brillante mucoide, con amplias áreas necróticas. En las zonas mejor
conservadas el tumor es sólido, blanquecino y muy friable.
Se realiza un amplio muestreo del tejido remitido y las numerosas preparaciones examinadas muestran una tumoración mesenquimal maligna heterogénea de diferenciación múltiple. El tumor es fundamentalmente sólido presentando tres patrones de crecimiento bien definidos. El primero (fig. 3) está formado por células fusiformes de núcleos alargados dispuestas en haces entrelazados irregulares de aspecto fibrosarcomatoso o leiomiosarcomatoso en sus áreas mejor diferenciadas; presenta, sin embargo, una variabilidad notable con focos de células pleomórficas laxas rodeadas de estroma mixoide (fig. 3, detalle), en otras el aspecto es lipoblástico con células redondeadas de citoplasma vacuolado y núcleo central. El segundo patrón (fig. 4) es más indiferenciado, con células de contorno irregular, escaso citoplasma, núcleos anisocarióticos pleomórficos, de tipo blastematoso y alto índice mitótico (fig. 4, detalle), que se disponen formando placas amplias o nidos dispersos entre los haces de tipo leiomiosarcomatoso. El tercer patrón es de tipo condroide (fig. 5) formando islotes dispersos en la masa tumoral. Su grado de diferenciación es variable, en ocasiones solo presenta un discreto esbozo, mientras que en otras áreas su aspecto es de cartílago hialino atípico. De forma parcheada el tumor pierde su aspecto sólido y presenta hendiduras o espacios microquísticos en los que se puede observar un revestimiento cilíndrico ciliado con células caliciformes de tipo respiratorio con focos de metaplasia escamosa (fig. 6-A) y debajo del mismo una capa continua de células blastematosas que remedan la capa de cambium de los rabdomiosarcomas botrioides (fig. 6-B). El tumor carece de componente epitelial maligno discernible. La superficie del mismo esta revestida por células cuboideas de aspecto mesotelial.
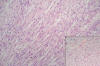
Fig. 3:
La imagen presenta el patrón predominante en el tumor. Células fusiformes
dispuestas en haces que recuerdan al fibrosarcoma o al leiomiosarcoma. Detalle:
en ocasiones y de forma salpicada se observaban áreas con células de mayor
tamaño, pleomórficas y rodeadas de una estroma laxo. (HE, 100x).

Fig. 4:
Focos blastematosos formados por células pequeñas primitivas malignas de núcleo
grande con alta relación núcleo/citoplasma que se distribuyen en grandes grupos
a través del tumor. Detalle: alto índice mitótico que superaba las 10 mitosis
por cada 10 campos de alto aumento (HE, 100x, 200x).

Fig. 5:
Áreas condroides que forman islotes dispersos a través de todo el tumor, se
puede observar la sutil transición-diferenciación que desde el tejido
blastematoso se produce hacia los islotes condroides. Detalle: en ocasiones los
islotes condroides se disponen alrededor de estructuras tubulares como bronquios
rudimentarios (HE, 100x, 200x).
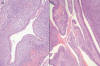
Fig. 6:
Las áreas quísticas presentan un revestimiento cilíndrico ciliado de tipo
respiratorio con metaplasia escamosa (A). En otras zonas, inmediatamente debajo
del epitelio de revestimiento se observa una capa de células blastematosas de
disposición parecida a la capa de cambium del rabdomiosarcoma botrioides (B),
(HE, 100x, 40x),
Inmunohistoquímicamente el tumor es desmina positivo (fig. 7) tanto en las zonas fasciculadas como en las de tipo cambium, siendo menos marcada la positividad en las zonas blastematosas (tinción de tipo Golgi/anillo perinuclear). Es interesante destacar que en las zonas fasciculadas se pueden observar algunas células con estriación transversal, positivas para actina de músculo estriado (fig. 7, detalle) que sugerían diferenciación rabdoide. Las áreas condroides mostraban positividad para S-100. El tumor no es reactivo para la actina de músculo liso, MyoD1, MIC-2, caldesmón o calponina. No presenta reactividad para CEA, a-fetoproteína o para marcadores neuroendocrinos (cromogranina, sinaptofisina y enolasa). La citoqueratina solo es positiva en las células de revestimiento mesotelial superficial y en el revestimiento de tipo respiratorio presente en las hendiduras o espacios microquísticos.

Fig. 7:
La imagen muestra tinción para desmina, predominante en el tumor. Detalle:
ilustra algunas células aisladas que presentaban positividad para actina de
músculo estriado con bandas citoplásmicas transversales (40x, 400x).
El índice mitótico del tumor supera las 10 mitosis por cada 10 campos de alto aumento y presenta un factor de proliferación Ki67 con una positividad del 25 al 60% según zonas.
Con estos resultados se diagnostica de blastoma pleuropulmonar de pulmón izquierdo.
DISCUSIÓN
El BPP es un tumor intratorácico maligno, primario de la infancia extremadamente raro, del que en la literatura mundial y en el Registro de Blastomas Pleuropulmonares (The Pleuropulmonary Blastoma Registry –www.ppbregistry.org–) se han descrito alrededor de 100 casos (18). No fue hasta 1988 que un grupo de tumores mesenquimales anaplásicos de la periferia del pulmón y de la pleura, en niños, se agrupan bajo el nombre de «blastomas pulmonares pediátricos» (14). Manivel y cols. (1) describen la entidad clínico-patológica, a partir de una serie de 11 casos de tumores pediátricos intratorácicos que comparten características comunes y que los diferencia del blastoma pulmonar del adulto. Esta neoplasia está encuadrada dentro de los tumores embrionarios o disontogénica que se observan, únicamente, en la infancia y entre los que se incluyen el neuroblastoma, hepatoblastoma, retinoblastoma o el tumor de Wilms (2,18).
Aproximadamente un 25% de los casos se acompañan de una predisposición constitucional o hereditaria a enfermedades neoplásicas o displásicas en consonancia con un síndrome canceroso familiar (12). En los niños afectados se ha observado nefromas quísticos, teratoma ováricos, pólipos intestinales múltiples o un segundo BPP (12).
Varios autores destacan la relación entre el BPP y cambios quísticos preexistentes, del tipo de la malformación quística adenomatoide, los secuestros pulmonares extralobares o los quistes boncogénicos, y sugiriendo un origen ontogénico común del BPP y estas entidades (2,6,8,19).
La edad de presentación oscila entre un mes y 12 años, siendo la media de 24 meses, con una incidencia similar en ambos sexos (18). Es interesante destacar una diferencia de edad de presentación, estadísticamente significativa, entre los tipos I –10 meses–, II –34 meses– y III –44 meses– (2). El caso que presentamos es fundamentalmente sólido y estaría dentro del margen de edad de los tumores tipo III.
La forma de presentación más común es con tos, disnea, fatiga y fiebre. A la exploración suele observarse derrame pleural y la sospecha clínica inicial suele ser de empiema o neumonía (6,14,18).
El lado derecho es la localización más frecuente, con un 64% de los casos en las series más largas, siendo el lóbulo inferior el comúnmente afectado (2). En un 40% de los casos se ve afectada también la pleura parietal (2). Hay descrito un solo caso de afectación bilateral (9).
Macroscópicamente son tumores blandos, friables y en buena parte de los casos necrosados, hasta tal punto que los cirujanos llegan a interpretar sus hallazgos intraoperatorios como un empiema (2). Sus dimensiones oscilan entre 2 y 28 cm de diámetro y pueden alcanzar hasta los 1.100 g de peso (2).
El cuadro histológico de estos tumores es muy polimorfo con presencia de células pequeñas y primitivas, de características blastematosas, separadas por estroma laxo y focos de diferenciación divergente rabdomiosarcomatosa, condrosarcomatosa, fibrosarcomatosa o liposarcomatosa, similar a lo observado en nuestro caso (1,2,3,15). La presencia de tejido epitelial, de aspecto benigno, en forma de revestimiento o islotes dentro de áreas mesenquimatosas son hallazgos frecuentes en este tumor y se interpretan como tejido atrapado por el crecimiento tumoral (2,7). El perfil IHQ es el de un sarcoma de alto grado reactivo para vimentina, actina, desmina y S-100 en los focos condroides.
Dada la extraordinaria variabilidad histológica de estos tumores, sobre todo en los sólidos (tipo III) el diagnóstico diferencial clínico abarca un amplio abanico de lesiones. Microscópicamente, el pseudotumor inflamatorio del pulmón, que es el tumor primario pulmonar más común en niños (8), debe ocupar el primer lugar y después el rabdomiosarcoma, el leiomiosarcomas, el histiocitoma fibroso maligno o el tumor maligno de vainas nerviosas periféricas. Todos estos tumores, sin embargo, presentan una diferenciación unidireccional, miógena, neuroide o fibroblastica, mientras que el BPP se caracteriza por una diferenciación multidireccional (3). No debe de olvidarse la posibilidad de los tumores de células germinales malignos, el tumor de Wilms extrarrenal, el mesotelioma maligno y las neoplasias tímicas malignas (2). Respecto al blastoma pulmonar, tumor que actualmente se encuadra dentro de los carcinomas sarcomatoides del pulmón, junto con el carcinoma sarcomatoide, el carcinoma de células fusiformes, el carcinoma de células gigantes y el carcinosarcoma (18); aunque comparte un cierto número de características comunes con el BPP: localización periférica, tendencia a la afectación de la pared pleural, presencia de un componente celular de tipo blastema y de estroma primitivo mesenquimal con ocasionales focos de osteosarcoma, condrosarcoma o rabdomiosarcoma (14,18); es, sin embargo, un tumor epitelial bifásico del adulto, que contiene una mezcla de células epiteliales tubulares malignas y grupos compactos de células fusiformes indiferenciadas de tipo blastema, en contraste con el BPP que es un tumor exclusivamente sarcomatoso, sin componente epitelial maligno. El blastoma pulmonar no presenta una tendencia particular a la asociación con otras enfermedades o a la agrupación familiar como ocurre en el BPP (14,12) y su perfil IHQ es el propio de un tumor epitelial: queratina, EMA y CEA positivos, marcadores que no están presentes en el BPP (18).
Entre las alteraciones genéticas que se observan en el BPP se han encontrado trisomías del cromosoma 2, también presentes en el rabdomiosarcoma embrionario (7) con el que comparte características clínicas y patológicas, lo que podría sugerir una génesis tumoral común. Novak y cols. (5) han informado así mismo de trisomías del cromosoma 8 en tres casos de BPP que incluían dos casos de tipo II (sólido-quístico) y un caso de tipo III (sólido).
El pronóstico del BPP es pobre en la mayor parte de los casos, los niños mueren dentro del primer año tras el diagnóstico. Se ha informado de supervivencias del 45% a los 5 años y únicamente del 8% a los 10 años (6). En la serie más extensa publicada por Priest y cols. (1997) se informa de recurrencias en el 50% de los casos y de metástasis en el SNC y médula espinal en el 44% de los casos, tendencia significativamente mayor que en otros tipos de neoplasias sólidas de la infancia. Debe indicarse que el seguimiento prolongado de los pacientes es muy recomendable ya que, en algunos casos, las metástasis en el SNC se produjeron hasta 60 meses después del diagnóstico. En la serie de Priest y cols. (1997), los pacientes con afectación mediastínica o pleural presentaban el peor pronóstico, siendo el tipo I (quístico) el mas fácilmente resecable y de mejor supervivencia a largo plazo.
Parece que existe una relación directa entre el tipo histológico (quístico –I–, sólido-quístico –II– y sólido –III–), la edad de aparición y la agresividad del BPP. Así los tipos I suelen ser más localizados, de menor tamaño, más fácilmente resecables y de mejor pronóstico a largo plazo, mientras que los tipos II y III son de agresividad creciente y se propone que las terapias complementarias (quimio- o radioterapia) deben aplicarse tan agresivamente como el paciente tolere (2).
Por último, respecto a las áreas revestidas por epitelio verdadero, se cita (2) que no aparecen en las metástasis, por lo que se considera que representan tejido pulmonar o pleural benigno atrapado por el tumor en su progresión. En nuestro caso, no tenemos datos del comportamiento del tumor en las metástasis ya que carecía de ellas al diagnóstico, pero la morfología de los restos epiteliales, regulares y sin atipias nos hacen coincidir con las apreciaciones de otros autores respecto a la probable benignidad de dichos restos. La presencia aislada de cambios escamosos o focos de hiperplasia mesotelial pensamos que son de naturaleza reactiva.
BIBLIOGRAFÍA
Manivel JC, Priest JR, Watterson J, Steiner M, Woods WG, Wick MR et. al. Pleuropulmonary blastoma: the so-called pulmonary blastoma of childhood. Cancer 1988; 62: 1516-26.
Priest JR, McDermott MB, Smita B, Watterson J, Manivel JC et. al. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer 1997; 80: 147-160.
Granata C, Gambini C, Carlini C et al. Pleuropulmonary blastoma: case report. Eur J Pediatr Surg 2001; 11: 271-3.
Wright JR. Pleuropulmonary blastoma: A case report documenting transition from type I (cystic) to tipe III (solid). Cancer 2000; 88: 2853-8.
Novak Robert, Sarada Dasu MD, Dimitris Agamanolis MD, et. al. Trisomy 8 is a characteristic finding in pleuropulmonary blastoma. Pediatric Pathol Lab Med 1997; 17: 99-103.
Hasiotou M, Polyviou P, et.al. Pleuropulmonary blastoma in the area of a previously diagnosed congenital lung cyst: Report of Two Cases. Acta Radiologica 2004: 289-92.
Yang Peng, Hasegawa Tadashi, et. al. Pleuropulmonary Blastoma: Fluorescence In Situ hibridization analysis indicating trisomy 2.Am J Surg Pathol 1997; 21: 854-9.
Lopez-Andrew Juan, Ferris-Tortajada Josep, Gomez Jacinto. Pleuropulmonary blastoma and congenital cystic malformations. J Pediatr 1996; 129: 773-4.
Picaud JC, Levrey H, Bouvier R, et. al. Bilateral cystic pleuropulmonary blastoma in early infancy. J Pediatr 2000; 136: 834-6.
Roque L, Rodrigues R, et. al Comparative genomic hybridization analysis of a pleuropulmonary blastoma. Cancer Genet Cytogenet 2004; 149: 58-62.
Hong B, Chen Z, et. al. Molecular cytogenetic analysis of a pleuropulmonary blastoma. Cancer Genet Cytogenet 2003; 142: 65-9.
Priest J, Watterson J, Strong L, et. al. Pleuropulmonary blastoma: A marker for familial desease. J Pediatr 1996; 128(2): 220-4.
Dehner LP, Watterson J, Priest J. Pleuropulmonary blastoma: A unique intrathoracic-pulmonary neoplasm of childhood. Perspect Pediatr Pathol 1995; 18: 214-26.
Leslie OK and Wick MR, editores. Practical Pulmonary Pathology. A diagnostic approach. Philadelphia: Churchill-Livingstone. 2004. p. 507-8.
Hachitanda Y, et. al. Pleuropulmonar blastoma in childhood. A tumor of divergent differentiation. Am J Surg Pathol 1993; 17: 382-91.
Romeo C, Impellizzeri P, et. al. Pleuropulmonary blastoma: long-term survival and literature review. Med Pediatr Oncol 1999; 33: 372-6.
Schmaltz C, Sauter S, et. al. Pleuro-pulmonary blastoma: a case report an review of the literature. Med Pediatr Oncol 1995; 25: 479-84.
Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, editors, World Health Organization classification of tumours. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon: IARC Press 2004: 99-100.
Valderrama E, Saluja G, Shende A, Lanzhowsky P, Berkman J. Pulmonary blastoma: report of two cases in children. Am J Surg Pathol 1978; 2: 415-22.
![]()