
Vol. 41, n.º 2, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 2, 2008 |
REVISIONES
Manuel Vaquero1
1 Hospital Donostia.
Paseo Doctor Begiristain. 20014 Donostia, San Sebastián.
mvaquero@chdo.osakidetza.net
RESUMEN
Las enfermedades intersticiales pulmonares constituyen un grupo heterogéneo de procesos con patogénesis y pronósticos muy diferentes. Las formas idiopáticas son un grupo importante de estos procesos intersticiales. Los hallazgos histológicos no son patognomónicos por lo que el patólogo debe entender cómo valorar correctamente los datos clínicos, las pruebas funcionales y la TAC de alta resolución, para realizar adecuadamente una aproximación al diagnóstico anatomopatológico. Se comentan las nuevas perspectivas sobre la patogénesis general y el tratamiento. También se revisan las enfermedades intersticiales idiopáticas hereditarias y los progresos alcanzados en su conocimiento.
Palabras clave: Neumonía intersticial idiopática, enfermedad intersticial hereditaria, patogénesis, fibrosis pulmonar.
SUMMARY
Interstitial lung diseases are an heterogeneous group of disorders with pathogenesis and prognosis very different. The idiopathic forms are an important group of these interstitial process. There is not pathognomonic histological signs and pathologists must get to know the appropriate interpretation of the clinical, functional and high-resolution CT to achieve a correct pathologic diagnosis. New insights into general pathogenesis and treatment are commented. A review about actual progress in the understanding of inherited interstitial lung diseases are also included.
Keywords: Idiopathic interstitial pneumonia, inherited lung disease, interstitial lung disease, pathogenesis, pulmonary fibrosis.
INTRODUCCIÓN
Las enfermedades pulmonares intersticiales engloban varios grupos de procesos (tabla 1) con una serie de hallazgos comunes (1,2): clínica de tos y/o disnea, infiltrados intersticiales difusos y alteraciones en la fisiología del intercambio de gases en reposo o durante el ejercicio. Morfológicamente hay grados variables de inflamación, fibrosis y remodelación tisular. Apenas conocemos la patogénesis de las neumonías intersticiales y sólo se identifica un agente causal en el 35% de los pacientes, por lo que la mayoría de los casos se consideran idiopáticos. A estos grupos habría que añadir las neumonías intersticiales propiamente pediátricas y su posible relación con la neumonía intersticial familiar (3-5), en los que hay afectación de al menos dos familiares de primer grado. La variabilidad clínica, radiológica y patológica es más frecuente que en los casos no familiares y la afectación es más intensa en los fumadores, en los de sexo masculino y de mayor edad. La prevalencia de las neumonías intersticiales idiopáticas (NII) del adulto se incrementa con la edad, variando entre 2,7/100.000 a los 35-40 años a más de 175/100.000 cumplidos los 75 (6).

Entre 1931 y 1943, Hamman y Rich recogen cuatro pacientes estudiados en el hospital John Hopkins que fallecieron en menos de seis meses por insuficiencia respiratoria. Realizaron una correlación clínico-radiológica-histológica de un síndrome que posteriormente se denominó con los nombres de estos autores y que actualmente podría reclasificarse como neumonía intersticial aguda (7). En 1957 Rubin y Lubiner revisan 39 casos publicados junto a 15 propios y describen «variantes» del síndrome de Hamman-Rich. Algunos de estos pacientes tenían artritis reumatoide. Posteriormente se describen casos «crónicos» del síndrome hasta que en 1969, Liebow y Carrington (8) proponen la primera clasificación de las neumonías intersticiales idiopáticas. De los seis tipos propuestos se descolgó la neumonía de células gigantes al demostrarse su asociación a vapores de tungsteno producidos durante el procesamiento de metales pesados, como el cobalto (6). Más de un cuarto de siglo después, Katzenstein y Myers (9,10) revisan la clasificación e introducen el término de neumonía intersticial no específica (NINE) donde incluyen un grupo de pacientes con características morfológicas diferentes a la de los grupos conocidos. Tiene, además, un curso clínico más favorable que la neumonía intersticial usual. En junio de 2001 se alcanza un consenso entre clínicos, radiólogos y patólogos de la American Thoracic Society y de la European Respiratory Society (ATS/ERS) sobre las definiciones y los criterios diagnósticos de las NII (11). El cambio más sustancial consiste en que la definición de cada entidad descansa en criterios clínicos, en la TAC de alta resolución (TACAR) y en los hallazgos anatomopatológicos. El diagnóstico final es por tanto multidisciplinar, lo que mejora las variaciones interobservador en el diagnóstico final. Esta clasificación sigue todavía en uso, en el adulto, y es la que vamos a comentar.
Utilidad de la TACAR en las neumonías intersticiales
La radiografía de tórax es normal en el 10 a 15% de las enfermedades intersticiales pulmonares (12). La TACAR (13,14) no sólo es importante en el diagnóstico de las enfermedades intersticiales pulmonares sino también en la selección de los lugares para biopsiar. A nivel diagnóstico las imágenes son muy características y pueden evitar la biopsia en la histiocitosis de células de Langerhans y en la linfangiomiomatosis. También puede evitar la toma de biopsias en pacientes con sarcoidosis y alveolitis alérgica con clínica compatible. Una TACAR compatible con neumonía intersticial linfocitaria (NIL) puede evitar una biopsia si hay antecedentes de síndrome de Sjögren y linfocitosis en el lavado bronquioloalveolar.
En 1 mm3 de parénquima pulmonar hay aproximadamente 170 alvéolos por lo que las técnicas de imagen no pueden distinguir entre espacio alveolar e intersticial (15). La unidad anatómica más pequeña visible en la TACAR es el lobulillo secundario de Miller, o sea la zona más pequeña de parénquima pulmonar delimitada por septos conectivos. Zompartori y col. hacen una revisión de los hallazgos de la TACAR en enfermedades pulmonares difusas primarias o secundarias (12). Los patrones que pueden encontrarse son: nodular, reticular, quístico, «vidrio deslustrado» y consolidación. El patrón nodular varía de 0,1 a 1 cm, y según las causas puede tener una distribución perilinfática, centrilobular o al azar (miliar). En el reticular los septos interlobulares están engrosados de forma lisa o irregular. Dentro del patrón quístico se incluyen, además de estructuras circunscritas de paredes finas y contenido aéreo o líquido, el de panalización. Este patrón muestra paredes más gruesas, bien definidas y de predominio subpleural y basal. Está presente en el 70% de las FPI. Se habla de «vidrio deslustrado» cuando se observan zonas de incremento de la atenuación pero que mantienen la visibilidad de las paredes bronquiales y vasculares. Se presenta si hay disminución relativa del aire o remplazamiento del mismo por otras sustancias. Suele observarse en casos con histología de bronquiolitis obliterante con neumonía obstructiva (BONO) en la que característicamente pueden emigrar y tiene una distribución bilateral, periférica y asimétrica. Se habla de «atenuación en mosaico» cuando aparecen áreas parcheadas bien definidas de incremento relativo y disminución de la atenuación pulmonar. Las imágenes en espiración sirven para diferenciar si este patrón se debe a enfermedad de pequeñas vías respiratorias o a enfermedad oclusiva pulmonar. El patrón en «vidrio deslustrado» es visible en el 80% de la NINE, generalmente bilateral, basal y subpleural. La consolidación se diferencia del anterior en que no se ven los vasos pero puede haber broncograma aéreo.
En cualquier caso el diagnóstico se mejora y afina más con el trabajo en grupo de clínicos, radiólogos y patólogos, alcanzándose el mismo en el 81% de los pacientes. En casi la mitad de los casos con FIP-NINE los hallazgos patológicos son críticos para el diagnóstico final, ya que tanto los clínicos como los radiólogos tienden a sobrediagnosticar NINE.
Cuando se haga indicación de TACAR en niños es importante considerar que la dosis de radiación que reciben es importante. Deben obtenerse imágenes inspiratorias y espiratorias, para detectar el atrapamiento de aire y la posible presencia de imágenes en «vidrio deslustrado». La seguridad diagnóstica de la TACAR en enfermedades difusas pediátricas es del 100% en la proteinosis alveolar y en la hemosiderosis idiopática, del 50% en la NID y del 33% en la bronquiolitis folicular y NIL (15).
Flaherty et al (16) revisaron 58 pacientes consecutivos con sospecha de neumonía intersticial idiopática. En 30 de ellos el diagnóstico de consenso fue de fibrosis pulmonar idiopática (FPI). Antes de la información morfológica los clínicos identificaron el 75% de los casos y los radiólogos el 48%. El mayor impacto de los datos anatomopatológicos sobre el diagnóstico final se produjo en los casos que no eran FPI, con rectificación del diagnóstico en el 54 al 71% de los pacientes (16). Sin embargo, el acierto clínico-radiológico fue elevado en los casos de fibrosis pulmonar idiopática, lo que apoya las recomendaciones previas del panel de expertos (11) que proponen obviar la biopsia quirúrgica en adultos inmunocompetentes que cumplan con determinados criterios clínico-radiológicos (tabla 2). Wells (17) también llega a las mismas conclusiones. En tan sólo 5 de 104 pacientes con FPI el diagnóstico clínico-radiológico fue discordante con la histología (valor predictivo >95%). Los mayores problemas se debieron al error de muestreo y a las variaciones interobservador. El error de muestreo es mayor en los casos donde debe diferenciarse una neumonía intersticial no específica (NINE) de la FPI pero puede disminuirse si el cirujano planifica la zona de biopsias mediante los hallazgos de la TACAR y cumple una serie de requisitos (18) (tabla 3). Las importantes variaciones interobservador (kappa 0,32-0,43) reflejan el gran número de casos con morfología intermedia entre ambas entidades (19-21). Flaherty y col. (16) analizan los resultados de 109 pacientes con sospecha de enfermedad pulmonar difusa y biopsias de varios lóbulos pulmonares. En 51 pacientes se diagnosticó FPI en todos los lóbulos y en 25 NINE, tan sólo cinco con patrón celular. El resto de pacientes (28%) tenían un patrón de FPI en al menos un lóbulo y NINE en los otros.


La biopsia pulmonar
La tendencia actual para la obtención de cuñas pulmonares, tanto en el adulto como en niños, es la videotoracoscopia (22) que tiene una morbilidad / mortalidad en pacientes sin «alto riesgo» del 10 y 1% respectivamente. Las complicaciones más frecuentes son: la pérdida de aire durante más de cinco días, recidiva del neumotórax y la hemorragia y/o dolor prolongado. Los pacientes considerados de «alto riesgo» son aquellos con insuficiencia respiratoria (pO2 menor de 50-60 mmHg), hipoxia extrema que requiere ventilación mecánica, hipertensión pulmonar, diátesis hemorrágica e inmunodepresión (22). La toracotomía estándar se recomienda en pacientes de alto riesgo y en aquellos con adherencias pleurales intensas.
La reunión internacional de consenso (ATS/ERS) se propuso como objetivo la estandarización de la clasificación de las neumonías intersticiales idiopáticas del adulto (11). Para ello, se revisaron tanto pacientes como la evidencia científica publicada en inglés (Medline 1966 a 1988) y se aceptaron las entidades clínico-radiológico-morfológicas que comentaremos a continuación. Para aquellos casos en que el patólogo no cuente con las aportaciones necesarias de los clínicos o radiólogos, Leslie (23) plantea una clasificación muy útil del proceso dentro de seis patrones morfológicos. En cada uno de ellos valora los diagnósticos diferenciales posibles según el tipo de células inflamatorias, las estructuras afectadas o si hay pleuritis asociada, así como por el tipo de fibrosis y otros datos anatomopatológicos.
Fibrosis pulmonar idiopática (FPI)
Entre el 40 y el 60% de pacientes adultos con enfermedad pulmonar intersticial difusa se corresponden con esta entidad (24). La prevalencia es de 13 y 20 casos/100.000 habitantes, en mujeres y hombres respectivamente (25). Afecta preferentemente a mayores de 50 años y en un 10-20% de los casos hay títulos bajos de anticuerpos antinucleares y de factor reumatoide. La imagen clásica en la TACAR es de reticulación subpleural bilateral con bronquiectasis por tracción y panalización (26). En el lavado bronquioloalveolar (LBA) predominan los polinucleares. La respuesta al tratamiento con corticoides con o sin azatioprina es incompleta y el pronóstico malo, con una supervivencia desde el inicio de los síntomas de 80,8 meses y de tan sólo 35,2 meses tras el diagnóstico (1,25-28).
La histología es de neumonía intersticial usual cuyo signo distintivo es la presencia de focos fibroblásticos –áreas discretas pero progresivas de proliferación fibroblástica en la periferia de los lóbulos– (fig. 1). Existen zonas del parénquima en distintos estadios de evolución y frecuente patrón «en panal» (23). La evolución temporal en brotes explicaría los distintos estadios evolutivos morfológicos en un mismo paciente (29). Katzentein y Myers (30), así como otros autores, encuentran correlación entre la cuantía de focos fibroblásticos y el pronóstico (7,31,32). Noble (33) recoge un score medio de focos fibroblásticos de 1,8 (percentil 25: 1, percentil 75: 2,7). El incremento de una unidad en la cifra de tanteo se asoció a un incremento del 74% en el riesgo de muerte. La utilización de métodos cuantitativos, del tipo de análisis de imagen, puede eliminar los problemas de cuantificación, tiempo y reproducibilidad del recuento de los focos fibroblásticos (34). Cool y col. (35), tras realizar morfometría en cortes seriados proponen que los focos fibroblásticos formarían un verdadero retículo, una «ola de fibrosis», que progresaría a través del pulmón. El diagnóstico diferencial morfológico debe hacerse fundamentalmente con (6): formas fibróticas de NINE, asbestosis, colagenosis (36-39), formas crónicas de neumonitis por hipersensibilidad y síndrome de Hermansky-Pudlak (fig. 2).

Fig. 1:
Fibrosis pulmonar idiopática en la que alternan
zonas casi normales con otras marcadamente fibróticas con metaplasia columnar
incipiente. La ausencia de arteria pulmonar lo diferencia de la vía aérea.
Presencia de focos de proliferación fibroblástica (recuadro).
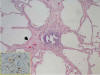
Fig. 2:
Histiocitosis de células de Langerhans en fase
fibrótica, simulando FPI. La inflamación evidente, la proximidad al centro del
acino (músculo liso organizado en bronquiolo: flecha) y la positividad del CD1a
(recuadro) ayudan a la diferenciación.
Para poder valorar las implicaciones que los conocimientos patogénicos tienen en los recientes y futuros tratamientos, vale la pena conocer con cierta profundidad las diversas alteraciones estudiadas. En las posibles patogénesis hay distintas épocas (7,40); en los años 70 se proponía una teoría inflamatoria a pesar de la ausencia de inflamación intersticial o alveolar, incluso en las fases precoces del proceso. En la década de los 80 cobran auge los factores del crecimiento, como el PDGF-beta y el TGF-beta, al demostrarse que los macrófagos del BAL de estos pacientes producían proliferación de fibroblastos en un medio de cultivo; así como la interrelación epitelial-mesenquimal (41). En los estudios ultraestructurales se observó apoptosis del epitelio alveolar y colapso del alveolo caracterizado por la invaginación de la lámina basal denudada que posteriormente se reepitelizaría.
En la fase de iniciación del proceso de la FPI se produciría la apoptosis de los neumocitos tipo II (42) y estaría promovida por sustancias oxidantes y por citocinas, como la IL-1 y el TNF-a, producidas por macrófagos activados (43,44). Se ha descrito la presencia de ADN de herpesvirus en los neumocitos, por lo que se plantea su posible rol en el inicio y/o mantenimiento de las lesiones (45).
En la patogénesis de la enfermedad pulmonar intersticial (46,47) (fig. 3) se cree que son fundamentales dos alteraciones. La asociada a la reparación llevada a cabo por los neumocitos tipo II –reconocibles por su positividad frente a la proteína A asociada al surfactante– y el remoldeamiento vascular En este último se producen anastomosis entre la microvasculatura pulmonar y sistémica (48), y se ha estudiado fundamentalmente en la fibrosis pulmonar idiopática (43). En la FPI la reepitelización de los espacios alveolares y del tejido neoformado intraluminal está disminuida. En esa ausencia efectiva de reepitelización parece clave la rotura de la membrana basal, sobre todo de la laminina-5 g2 (44) presente en las membranas basales –para promover la unión del epitelio a la membrana basal– y en los propios neumocitos tipo II que están emigrando. El daño epitelial libera diversos factores de crecimiento entre los que destaca el TGF beta que promueve la fibrogénesis al incrementar la síntesis de colágeno y fibronectina por los miofibroblastos e inhibir su degradación (43). Además, el TNF beta tiene un efecto antiapoptótico sobre los miofibroblastos que parece ser importante en la patogénesis de la FPI (47,48). Los miofibroblastos podrían tener dos orígenes, bien de células epiteliales del pulmón bien de fibrocitos circulantes derivados de la médula ósea (49).
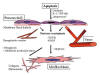
Fig. 3:
Patogénesis de la fibrosis pulmonar idiopática.
En cuanto a la remodelación vascular en la FPI hay datos aparentemente contradictorios (50). Mientras en las lesiones iniciales no fibróticas hay aumento del número de capilares (reconocibles por su endotelio CD34+), con un aumento de su proliferación; en las zonas fibróticas se produce una disminución progresiva de capilares hasta su desaparición y quedan únicamente reconocibles las vénulas poscapilares (por su endotelio CD34 negativo pero positivo con el factor de von Willebrand). Como factores implicados en estos hechos se ha encontrado una disfunción entre el factor de crecimiento del endotelio vascular (VEGF) y el factor derivado del epitelio pigmentado ocular (PEDF que regula el desarrollo vascular intraocular). La producción de este último parece regularse por el TGF-b1 que se encuentra aumentado en el epitelio que reviste los focos fibroblásticos (51). Otros inhibidores del remoldeamiento vascular serían la angiostatina y la quimocinas inducibles por IFN-gamma (49).
La matriz extracelular constituye el 25% del peso seco del pulmón y está constituida por colágeno en su 70%; normalmente de los tipos I y III en relación 2:1. En la fibrosis pulmonar la relación incrementa 3 ó 4 a 1 y, además, hay colágeno tipo IV y V en los espacios aéreos posiblemente como resultado de la rotura de la lámina basal que lo limita al compartimento intersticial. Actualmente se cree que es importante una interacción epitelial-fibroblástica y una acción de las proteinasas adecuadas, como las: serina, metalo, cisteína y aspártico proteinasas (52). Estas enzimas son fundamentales durante el desarrollo para la broncolización, alveolización y vascularización pulmonar. En la degradación de la matriz las más importantes son las metaloproteinasas. Por ejemplo la MMP-9, que por una parte rompería el colágeno tipo IV y, por otra, activaría el TGF-b que promueve la fibrosis e inhibe la diferenciación celular suprimiendo la expresión de la proteína B del surfactante. En las neumonías intersticiales, los inductores de las metaloproteinasas de matriz (MMP) extracelulares se encuentran en los neumocitos tipo II hiperplásicos y podrían ser importantes en la reepitelización (53).
Dos sistemas de receptores, tipo tirosincinasas, importantes en la remodelación tisular son el del EGFR y el PDGFR (54). El primero actúa sobre el epitelio y células mesenquimales subyacentes induciendo metaplasia mucinosa, hiperplasia y diferenciación mesenquimal y producción de matriz extracelular; mientras el segundo se localiza en células mesenquimales induciendo señales de supervivencia, crecimiento y quimiotáxis.
Zuo y col. (49) han descrito cuatro categorías de genes implicados en la regulación de la fibrosis pulmonar: 1. Los asociados a contracción celular, como la actina, miosina y tropomiosina; 2. Los asociados a proteínas de señalización, formación de matriz –colágeno tipos I y III– y degradación de la misma, como determinadas metaloproteinasas; 3. Aquellos relacionados con citocinas proinflamatorias, quimiocinas angiogénicas –tipo CXC (citocina con dos cisteínas separadas)– y antioxidantes; y 4. Los genes de las inmunoglobulinas.
Por último, pueden haber en estos pacientes alteraciones de los mecanismos endógenos de la reparación, como disfunción del IFN-g1b (55) –importante en la respuesta a infecciones y en la vigilancia antitumoral– y de la prostaglandina PGE2 que estaría disminuida en los fibroblastos de la FPI (56). El curso de la enfermedad parece asociado a exacerbaciones que acelerarían el declive de la función pulmonar (49).
En cuanto al tratamiento, en la revisión de ensayos clínicos randomizados (57-60) que utilizaron corticoides para el tratamiento de la FPI no se observó una eficacia significativa, por lo que se sugiere intentarlo con sustancias inmunomoduladoras. En un estudio multinacional con participación de 58 centros de EEUU, Canadá, Europa y Sudáfrica (58-60), 330 pacientes con FPI que no habían respondido previamente a corticoides se trataron con IFN-g1b o placebo (estudio GIPF-001). La elección del interferón se debió a sus propiedades antifibróticas, antiproliferativas e inmunorreguladoras. Por ejemplo, el IFN-gamma induce ITAC (IFN-inducible T-cell alpha chemoattractant) que, en ratones con fibrosis inducida por bleomicina, disminuye el depósito de colágeno tipos I y III (61). En el citado estudio clínico no se observaron cambios significativos en la duración de la supervivencia libre de progresión aunque hubo una tendencia a disminuir la mortalidad por el proceso, al reducirse el riesgo de fallecimiento un 9% sobre todo en los grupos con FVC y DLco menos alterados. Parece importante que en futuros ensayos deben refinarse las herramientas clínicas que permitan monitorizar y predecir mejor el curso de la enfermedad; una de las propuestas sería la realización del test de marcha durante seis minutos (58). Martinez (40) y Nathan (63) hacen sendas revisiones de ensayos recientes y la fase en que se encuentran, como el denominado INSPIRE (Internacional Study of Survival Outcome in Idiopahic Pulmonary Fibrosis) que finalizará en 2008 y valorará la supervivencia en unos 800 pacientes tratados con IFN-g1b. También recogen los resultados de otros tratamientos como el pirfenidone, antiinflamatorio, antioxidante y antifibrótico; el etanercept, antagonista del TNF-alfa, y la N-acetilcisteína. La combinación de esteroides con anticoagulantes parece mejorar la supervivencia durante las exacerbaciones. Asimismo es importante el tratamiento de la hipertensión pulmonar asociada.
La mayoría de los pacientes con FPI fallecen antes de los cinco años desde la realización del diagnóstico, y el trasplante pulmonar puede ser su única esperanza (63). Las líneas generales para el trasplante pulmonar en pacientes con FPI y NINE se recogen en la tabla 5. Es importante definir unos «predictores» de mortalidad que faciliten la selección de los pacientes para el trasplante (tabla 6). Wells y col. (64) proponen el CPI (Composite Physiologic Index) derivado de las mediciones de FEV 1, FVC y DLco como el mejor para predecir la mortalidad. La tasa de supervivencia postrasplante en FPI alcanza el 40% (65). Los riesgos de mortalidad temprana postrasplante se deben fundamentalmente al fracaso primario del injerto y a infecciones, mientras en la tardía suele ser, junto a las infecciones, el rechazo crónico (66).


Lynch et al (67) realizaron un estudio con 315 pacientes diagnosticados de FPI y seguidos durantes 58 semanas. El análisis estadístico multivariante identificó tres marcadores independientes de la mortalidad: el score de la fibrosis en la TACAR –combinación de la severidad de la reticulación y del patrón «en panal»–, el grado de afectación de la capacidad de difusión del monóxido de carbono y el tratamiento con IFN-gamma 1b (vesus placebo). Es una aportación importante (68) en la que además hubo un grado de correlación entre los radiólogos locales y los radiólogos expertos del 86%. El patrón de panalización estuvo presente en el 91% de los pacientes aunque no fue reconocido en bastantes ocasiones por los radiólogos locales. Por ello proponen la creación de unas imágenes de TACAR estandarizadas semejantes a las utilizadas por la Internacional Labour Organization para la valoración de la enfermedad pulmonar ocupacional (69).
Se ha intentado encontrar marcadores séricos y/o en el BAL que estén incrementados y que puedan utilizarse como indicadores pronósticos (70). La mayoría de ellos están implicados en la patogénesis de la FPI y son producidos sobre todo por neumocitos tipo II alterados (KL-6) y macrófagos alveolares (TGF-beta). La determinación en suero de la glucoproteína KL-6 ha resultado útil, como indicador de afectación pulmonar y progresión, en la esclerodermia (38) y dermatomiositis (37). Aunque en esta última el factor predictivo más importante de enfermedad intersticial pulmonar (37) es la presencia en suero de anticuerpos frente a la sintetasa aminoacidil-tRNA (anti-Jo 1). En cualquier caso, Raghu y Brown recogen los test más útiles, en sangre y orina, para el diagnóstico de la enfermedades intersticiales pulmonares (2).
Neumonía intersticial no específica (NINE)
No hay certeza sobre si la neumonía intersticial no específica es una entidad en sí misma (11) pero su reconocimiento y separación de la FPI ha tenido un impacto importante en el conocimiento de las neumonías intersticiales idiopáticas (17,27) tras su descripción por Katzenstein y Fiorelli en 1994. Corresponde al 14-36% de la FIP diagnosticadas antes de considerarse separadamente de la FPI. Un 39% de los casos son idiopáticos, mientras que puede ser simulado fundamentalmente por la esclerosis sistémica (38,39), la dermatomiositis (37), la neumonitis por hipersensibilidad (71), ciertos fármacos y la infección por VIH (72). La mitad de los pacientes presentan síntomas sistémicos: astenia, fiebre, disminución de peso y, hasta un tercio, acropaquias. El LBA es variable, con linfocitosis en la mitad de los casos y cociente T4/T8 disminuido. Las pruebas de función respiratoria son normales en el 14% de los pacientes, con patrón restrictivo puro en prácticamente el resto (72).
En la TACAR no hay panalización, y predominan las imágenes en «vidrio deslustrado», generalmente bilaterales, con distribución subpleural (73). La seguridad diagnóstica que aporta para diferenciarla de la FPI es del 66%, con una sensibilidad del 63% y una especificidad del 70% (74). Además, no hay diferencias significativas entre radiólogos locales y radiólogos expertos en esta patología, si ambos utilizan criterios predefinidos (74). Un patrón reticular más tosco y una menor proporción de atenuación en «vidrio deslustrado» favorecen más el diagnóstico de FPI (75).
A nivel microscópico las lesiones se encuentran en un mismo estadio. Se han descrito una fase o forma celular y otra con fibrosis (10). Si se ven áreas de neumonía intersticial usual se clasificará como fibrosis pulmonar idiopática. La discrepancia diagnóstica entre patólogos para diferenciarla de la FPI es del 52% (75).
El estudio del perfil genómico de las neumonías intersticiales (76,77) sitúa a la NINE superpuesta entre los genes presentes en la neumonitis por hipersensibilidad –asociados a inflamación e inmunidad– y los de la FPI –relacionados, sobre todo, con la remodelación tisular–.
Muchos pacientes, sobre todo los que tienen la forma celular, responden al tratamiento con corticoides (1). El pronóstico en conjunto, una vez ajustado sexo, edad y adicción al tabaco, es entre 17 y 24 veces mejor que el de la fibrosis intersticial idiopática (8). La supervivencia a los 10 años en la forma celular alcanza el 100%. La forma fibrosante tiene una supervivencia a los 5 y 10 años de 90 y 35% respectivamente frente al 43 y 15% de la FPI (72,78).
Bronquiolitis respiratoria asociada a enfermedad pulmonar intersticial (BR-EPI)
La bronquiolitis respiratoria es muy frecuente en los fumadores (>30 paquetes/año) pero no produce síntomas. En 1987 Myers (79) describe seis pacientes con bronquiolitis respiratoria morfológicamente –cúmulos de macrófagos pigmentados en espacios peribronquiolares- y con manifestaciones clínico-radiológicas de enfermedad pulmonar intersticial difusa– (1,28). La TACAR puede mostrar escasas imágenes en «vidrio deslustrado» y micromódulos centrlobulares mal definidos, sin panalización (80).
El proceso no progresa a fibrosis y regresa al dejar el hábito tabáquico (1). Algunos sujetos seguidos mediante LAB seriados, una vez han dejado dicho hábito, tardaron hasta tres años en normalizar sus niveles de macrófagos pigmentados (80).
Neumonía intersticial descamativa (NID)
Parece estar relacionada con la entidad precedente aunque no hay evidencia de evolución desde una BR-EPI (80). No se asocia a síntomas sistémicos ni, generalmente, a otras enfermedades. En la TACAR predominan las imágenes de atenuación en «vidrio deslustrado» aunque puede haber engrosamiento difuso de los septos alveolares (28,80).
Morfológicamente los alvéolos están rellenos de macrófagos con un fino pigmento granular parduzco (Perls positivo y, a veces, PAS positivo), mientras los septos alveolares no muestran prácticamente alteraciones. Un aspecto semejante puede verse en la periferia de lesiones ocupantes de espacio, así como en enfermedades pulmonares difusas (9): hemorragia alveolar, asbestosis, coniosis inorgánicas, proteinosis alveolar y neumonía eosinófila crónica. La abundancia de histiocitos gigantes multinucleados obliga a hacer el diagnóstico diferencial con patología por metales pesados (figs. 4 y 5).

Fig. 4:
Bronquiolitis respiratoria: zona centro-acinar
con espacios repletos de histiocitos pigmentados característicos (recuadro).

Fig. 5:
Neumonía descamativa por metales pesados
(cobalto) con las típicas células gigantes multinucleadas.
La supervivencia alcanza el 70% a los 10 años y suele responder al cese del consumo de tabaco y al tratamiento con corticoides (1).
Neumonía intersticial aguda (NIA)
Descrita por Hamman y Rich en 1935, tiene una presentación aguda y un curso fulminante. La TACAR muestra desde imágenes en «vidrio deslustrado» a consolidación confluente y difusa de los espacios aéreos. Se corresponde morfológicamente con el daño alveolar difuso de un distrés respiratorio (28). El lavado bronquiolo-alveolar muestra polinucleares y puede ayudar en el diagnóstico diferencial inicial con procesos como la neumonía eosinófila aguda o las neumonitis asociadas a colagenosis, medicamentos o hipersensibilidad (81). Es el segundo patrón morfológico más frecuente en la dermatomiosistis (37). Además de otras causas de distrés respiratorio agudo, debe descartarse una agudización de una fibrosis pulmonar idiopática.
Al inicio del proceso hay una fase exudativa, con presencia de membranas hialinas. Después de una o dos semanas se produce una organización del exudado intraalveolar tras la ruptura de la membrana basal alveolar y la migración y proliferación de fibroblastos y miofibroblastos, que puede conllevar una fibrosis intersticial con zonas de panalización y metaplasia escamosa y bronquiolar. Son frecuentes los trombos en arterias pulmonares. En la fase exudativa es fundamental el daño irreversible de los neumocitos y la liberación de mediadores inflamatorios –TNF-a, IL-1b– y de citocinas (81). La pérdida de la barrera epitelial motiva la exudación proteinácea a los espacios alveolares y la formación de las membranas hialinas.
La mortalidad en la fase aguda alcanza el 70%. El 40% de los sobrevivientes evolucionan a fibrosis, mientras otros recobran totalmente la función pulmonar o quedan con déficit funcional estable (81). Las recurrencias son raras. Los corticoides parecen ser efectivos en la fase exudativa (1).
En el año 2002 Beasley et al (82) describieron una neumonía aguda o subaguda que no cumplía los requisitos del daño alveolar agudo, de la bronquiolitis obliterante ni de la neumonía eosinofílica. Con el nombre de neumonía fibrinoide y organizada aguda reconocieron una posible variación del daño alveolar difuso con un patrón de daño pulmonar con los espacios alveolares repletos de cúmulos fibrinoides en forma de «bolas» pero sin membranas hialinas. La distribución es parcheada con leve engrosamiento de los tabiques alveolares con escasos linfocitos. Puede haber pequeñas áreas de bronquiolitis obliterante. La forma aguda tiene un curso fulminante. Más de la mitad de los casos se asociaron a enfermedades del colágeno, exposición a animales o a laca.
Neumonía organizada criptogenética (NOC)
La NOC (83) suele tener un inicio subagudo con tos, disnea y, a menudo, fiebre, astenia y disminución de peso. Evoluciona en semanas o meses y su pronóstico es favorable. Las pruebas de función respiratoria muestran un patrón restrictivo leve a moderado. En los casos típicos la TACAR evidencia predominantemente consolidación de espacios aéreos de distribución subpleural o peribronquiolar, de predominio basal, que pueden migrar (28). En otras ocasiones puede haber nódulos en relación con focos de neumonía organizada y también líneas irregulares por la posible inflamación (84). Hay casos con localización en lóbulos superiores que pueden cavitarse y confundirse con tuberculosis (85). En el LBA hay linfocitosis del 20 al 40% con disminución del cociente T4/T8 (84), a veces con neutrofilia y/o eosinofilia (menor del 25%). En el 31-44% de los pacientes (85) se asocia a otras condiciones (tabla 4).

El cuadro morfológico es el de una reparación tisular inespecífica con ocupación de espacios aéreos distales por yemas de tejido fibroinflamatorio laxo, conocidos como cuerpos de Masson (fig. 6). Este tejido se extiende de un alveolo a otro a través de los poros de Kohn ocasionando una distribución de la afectación parcheada en torno a pequeñas vías aéreas. Lo que se ha venido denominando morfológicamente BONO (bronquiolitis obliterante con neumonía organizada). Deben buscarse microabscesos de eosinófilos y microgranulomas para descartar una neumonía eosinófila y una neumonitis por hipersensibilidad respectivamente (figs. 7 y 8).
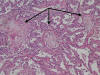
Fig. 6:
Cuerpos de Masson (flechas) en una neumonía
organizada criptogenética.
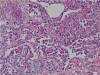
Fig. 7:
Neumonía eosinofílica crónica simulando una NID.
Obsérvense los abundantes eosinófilos y la formación de microabscesos
(recuadro).

Fig. 8:
Neumonitis alérgica extrínseca: la presencia de
microgranulomas y el predominio centroacinar ayudan en el diagnóstico
diferencial con la NINE celular. Recuadro: microgranuloma. El tamaño es muchas
veces menor.
En la patogénesis (84) se invoca a una estimulación inmunológica por antígenos desconocidos que se concentran sobre todo en bronquiolos distales y que dañan el epitelio. También participaría el tejido linfoide asociado a bronquio (BALT) junto al tejido de granulación de los cuerpos de Masson, indispensables para la reepitelización y restauración de la membrana basal. Posteriormente, un balance adecuado entre las metaloproteinasas y sus inhibidores titulares favorecería la reabsorción del tejido de granulación.
La evolución de la NOC es buena, con respuesta rápida a los corticoides en el 80% de los pacientes, aunque la recidiva es habitual al disminuir la dosis por lo que el tratamiento puede prolongarse bastantes meses (84). Las recidivas no alteran el pronóstico (1). Hay formas rápidamente progresivas que no responden a esteroides o, incluso, fulminantes que en bastantes casos –con un muestreo adecuado– pueden reclasificarse como NINE. Otras veces la evolución rápida se debe a que la enfermedad de fondo es grave o a que el paciente tiene patología importante asociada al tabaco.
Neumonía intersticial linfoide (NIL)
Se la considera actualmente una variante de la hiperplasia linfoide pulmonar con predominio de las alteraciones intersticiales (28). La forma idiopática es excepcional y deben descartarse sobre todo inmunodeficiencias (VIH) y enfermedades autoinmunes (Sjögren). Es un criterio para definir el sida pediátrico. El inicio suele ser subagudo con tos, disnea de esfuerzo y, en ocasiones, síntomas sistémicos con frecuente disglobulinemia. En el LBA hay linfocitosis evidente. La TACAR evidencia un patrón en «vídrio deslustrado» y, a veces, condensación y patrón micronodular (11).
A nivel microscópico hay un infiltrado linfocitario polimorfo exquisitamente intersticial, difuso y multifocal (fig. 9). A diferencia del linfoma no hay lesiones linfoepiteliales, destrucción alveolar ni afectación pleural (28). El 70% de los pacientes responden a los corticoides, mientras un 5% evolucionan a linfoma por lo que puede ser útil el estudio de la monoclonalidad de los linfocitos (1).
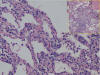
Fig. 9:
Neumonía intersticial linfoide. Infiltrados
linfocitarios difusos afectando estrictamente a los tabiques alveolares
(recuadro) y de mayor cuantía que los esperados en una NINE.
Enfermedad intersticial pulmonar pediátrica
En los niños las enfermedades intersticiales pulmonares son mucho menos frecuentes, con una prevalencia, desde el nacimiento hasta los 16 años, de 0,36/100.000. En la infancia es difícil de definir la enfermedad pulmonar intersticial dada la gran diversidad de procesos causales. Podríamos recogerlas en un síndrome con taquipnea, crepitantes, hipoxemia y/o infiltrados difusos. En la última década se han identificado nuevas entidades únicamente infantiles, algunas familiares unidas a defectos congénitos del surfactante.
Fan y col. (86) recogen como cuadros de enfermedad intersticial pulmonar únicos de la infancia los siguientes:
Taquipnea persistente infantil/hyperplasia de células neuroendocrinas de la infancia.
Bronquitis/bronquiolitis folicular.
Glucogenosis intersticial/neumonitis intersticial celular.
Hemorragia aguda idiopática pulmonar infantil.
Neumonitis crónica infantil/defectos genéticos de la función del surfactante.
La taquipnea persistente infantil (15,86) aparece durante el primer año de vida y muestra hiperinsuflación pulmonar en la radiografía de tórax, patrón en «vidrio deslustrado en la TACAR y signos de hipoxemia con atrapamiento de aire en las pruebas de función respiratoria. En la biopsia se evidencia una hiperplasia moderada del músculo liso peribronquiolar con aumento de macrófagos alveolares. Un hecho significativo es la hiperplasia de células claras neuroendocrinas en vías aéreas distales, puesta de manifiesto con inmunohistoquímica para bombesina, y microagregados en el parénquima, conocidos como cuerpos neuroendocrinos. Los síntomas pueden durar meses o años pero van mejorando con el tiempo sin que dejen secuelas en la vida adulta.
Otra entidad, con muy pocos casos reportados y que algunos autores relacionan con la anterior, es la bronquitis-bronquiolitis folicular en la que los infiltrados linfoides rodean y, focalmente infiltran, las vías aéreas. Se considera una hiperplasia del MALT bronquial y deben de descartarse alteraciones del sistema inmunológico (15,86).
Hace más de una década Schroeder, Shannon y Mark describieron la neumonitis intersticial celular infantil (87) en la que existe una proliferación intersticial de células con hábito monocitoide y ausencia de inflamación y fibrosis. Posteriormente (15,86), se han descrito monopartículas de glucógeno en estas células mesenquimales del intersticio que sugerirían una alteración en su citodiferenciación. Las pruebas de imagen muestran al inicio un patrón en «vidrio deslustrado» que progresa rápidamente a otro intersticial tosco (15). La evolución es buena, con una mortalidad del 20% en la docena de casos estudiados.
La neumonitis crónica infantil fue descrita por Katzenstein y col. (89) en nueve pacientes con engrosamiento septal, marcada hiperplasia neumocitaria y exudados alveolares de macrófagos. Es probable que tengan relación con anomalías genéticas de la función del surfactante ya que esta morfología se ha descrito asociada a mutaciones de SP-C (15,86).
El surfactante es una mezcla de fosfolípidos (80%), lípidos y proteínas específicas necesarios para reducir la tensión superficial del alveolo, lo que impediría su colapso al final de la espiración. El surfactante se produce en los neumocitos tipo II, en los cuerpos lamelares, segregándose por exocitosis. Además del déficit de surfactante por prematuridad, hay recién nacidos a término y niños mayores con enfermedad pulmonar secundaria a alteraciones en el metabolismo del surfactante. Las proteínas del surfactante pueden ser hidrofílicas, como la SP-A y SP-D, o hidrofóbicas como la SP-B y SP-C. Las primeras son esenciales para la defensa inmunológica y en la modulación de la respuesta alérgica del pulmón, mientras las hidrofóbicas reaccionan con los lípidos del surfactante para facilitar su adsorción a la interfase líquido-gaseosa (90). Las SP hidrofílicas son colectinas con cuatro dominios estructurales que incluyen un dominio colágeno y otro C-terminal. Este último, reconoce carbohidratos y media la interacción entre los patógenos y células apoptóticas y la fagocitosis, tras unirse a los receptores de superficie de los macrófagos alveolares y neumocitos tipo II (91). Los niveles de SP-D están disminuidos en la fibrosis quística y en el enfisema asociado al tabaco. Sus niveles séricos también disminuyen en las enfermedades intersticiales pulmonares por lo que se ha propuesto como un posible marcador «bronquioloalveolar» (92).
La proteína SP-B se codifica en el brazo corto del cromosoma 2 y cuenta con 11 exones. Los corticoides facilitan su transcripción pero mientras los neumocitos tipo II son capaces de procesar totalmente el proSP-B hasta el funcionalmente activo SP-B, no ocurre lo mismo en las células de Clara (93). El rol de la SP-B probablemente tenga relación con el empaquetamiento de los fosfolípidos en los cuerpos lamelares y es esencial en la proteolisis de proSP-C (94). La deficiencia de SP-B fue la primera causa genética conocida del síndrome de distrés respiratorio (SDR) y la mutación del codón 121 es la más frecuente, aunque suele ser única para cada familia afectada. La sintomatología aparece en las primeras 48 horas de vida y las técnicas de imagen muestran un patrón en «vidrio deslustrado», como una enfermedad por membrana hialina. Microscópicamente muestra una proteinosis alveolar con un material eosinófilo PAS-diastasa positivo en los alvéolos –posiblemente proSP-C aberrantemente procesada– y descamación de neumocitos con abundantes macrófagos. El paciente fallece por insuficiencia respiratoria a los 3-6 meses sin que se obtenga respuesta con SP-B exógena (94).
El gen de la proteína SP-C tiene 6 exones que transcriben la proteína proSP-C y que junto a la SP-B, necesaria para su proteolisis, es transportada a cuerpos multivesiculares y almacenada en los cuerpos lamelares una vez procesada. La proteína SP-C no está implicada en la síntesis ni en la secreción del surfactante, sino en la estabilización de los fosfolípidos del mismo. Además, juega un rol en la inmunidad innata pulmonar al alterar la unión de los lipopolisacaridos bacterianos inhalados al CD14 de los macrófagos que no liberarían tanta citocina TNF-a proinflamatoria (95). La enfermedad pulmonar asociada a SP-C puede deberse a la pérdida de proteína madura y/o a la acumulación de proSP-C tóxica en los neumocitos tipo II, al plegarse anómalamente en forma de estructuras b. Las mutaciones del gen SP-C puede clasificarse según se localicen en el dominio BRICHOS (dominio de unos 100 a.a. conservados en varias proteínas no relacionadas, como la condromodulina-1 asociada a condrosarcoma o la CA-11 asociada a cáncer gástrico) o estén fuera del mismo. Las primeras conducen a la formación de agregados perinucleares tóxicos –son más severas clínicamente– y las segundas inhiben el reciclaje del surfactante al acumularse. El aspecto morfológico también varía desde cambios parecidos a los de una proteinosis alveolar en el lactante hasta los más tardíos, sintomáticos en la vida adulta, que parecen una NINE celular o una FPI (94). Ultraestructuralmente los cuerpos lamelares son normales. La microscopía electrónica también es importante en las enfermedades de depóstico que afectan al pulmón (95).
Recientemente, se han descrito mutaciones de un gen localizado en el brazo corto del cromosoma 16 gen que codifica para la ATP-binding cassette protein A3 (ABCA3), situada en la membrana limitante de los cuerpos lamelares y, posiblemente, importante para su maduración y en la producción de surfactante (91,93,94). Los genes ABC codifican para proteínas implicadas en el transporte de compuestos a través de membranas. Hay 14 genes ABC asociados a enfermedades genéticas humanas, por ejemplo el ABCA1 se encuentra mutado en la enfermedad de Tangier por lo que se acumula colesterol en los macrófagos y tejidos periféricos con una deficiencia de lipoproteínas de alta densidad (94).
Bullard. (94) estudia 197 niños con enfermedad pulmonar difusa de causa desconocida y encuentra mutaciones de los genes SP-C y SP-B en 14,4% y 1% de ellos respectivamente. Del resto de pacientes hay un grupo de diez con mutación del gen ABCA3 (E292V), de los que casi un tercio son mayores de 10 años, nacidos de gestaciones a término, con síntomas respiratorios desde el primer año de edad y que tienen histología de NID en la biopsia. Por su parte Sherlenin y col. (95) secuencian el ADN sanguíneo para exones codificantes de surfactante en 337 niños, nacidos tras gestación de más de 36 semanas, con fallo respiratorio persistente iniciado a las pocas horas del nacimiento por déficit neonatal severo de surfactante. Encuentran 37 con déficit hereditario de la proteína B del surfactante, 6 con mutaciones del gen de la proteína B y, en un grupo de 21 lactantes con historia familiar y/o consanguinidad, 16 (76%) con mutaciones del gen ABCA3 que muestran imágenes muy anormales de los cuerpos lamelares: más pequeños y densamente empaquetados, así como con inclusiones periféricas más electrón-densas que les dan un aspecto en «huevo frito» y que parecen característicos de esta entidad (95). Las biopsias mostraron NID o proteinosis alveolar. Las mutaciones fueron diferentes según las familias, lo que sugiere que no hay un único alelo que confiera la enfermedad. Un paciente con mutación missense (Q1591P) en un alelo y desconocida en el otro, vive a los seis años con enfermedad pulmonar crónica. Esto sugiere que algunas mutaciones ABCA3 no son fatales. Dada la imposibilidad de realizar un cribado de todas las posibles mutaciones de las citadas en los sujetos con sospecha de padecer una enfermedad pulmonar difusa, se recomienda al menos la búsqueda de la mutación E292V, junto a la inmunohistoquímica de las proteínas del surfactante en la biopsia y al estudio de los cuerpos lamelares por microscopía electrónica (95,96).
Otros genes implicados en la morfogénesis pulmonar, expresión de las proteínas del surfactante o en la síntesis de los cuerpos lamelares son los siguientes (92): TTF-1, SNAP 23 y HPS 1-6. El TTF-1 es importante en la regulación de las proteínas del surfactante y su alteración, junto a distrés respiratorio, causa corea e hipotiroidismo. El SNAP 23 se necesita para la fusión de los cuerpos lamelares a la membrana plasmática de los neumocitos tipo II y sus alteraciones se asociarían más bien a malformaciones pulmonares letales. Los genes HPS están implicados en la biogénesis de lisosomas y organelas relacionadas. Están alterados en el síndrome de Hermansky-Pudlak, autosómico recesivo, que cursa con hipopigmentación, alteraciones de la coagulación y fibrosis pulmonar parecida a una FPI. Las alteraciones pulmonares se deben al impedimento de la secreción del surfactante y se acompaña de cuerpos lamelares anormales (95). Las alteraciones más graves son las asociadas a los genes HPS 1 y 4.
En la infancia no habría ni verdadera FPI ni bronquiolitis respiratoria, tal como las entendemos en el adulto. En el centenar de casos de FPI descritos en niños no constan los focos fibroblásticos tan característicos de este proceso y, además, el curso es mucho menos agresivo (86). La imagen morfológica más frecuente es la de NID, sobre todo durante el primer año de vida, aunque tiene mucho peor pronóstico que la del adulto (52). Debe descartarse que no se asocie a enfermedades sistémicas por depósito –Gaucher y Niemann-Pick (97)– o del colágeno, como la artritis reumatoide infantil o el lupus (15). La NIL pediátrica afecta a sujetos con VIH (15,98). Mientras que ante un diagnóstico de COP infantil, debe descartarse una fibrosis quística de fondo (86). No hay casos infantiles reportados de NIA por lo que debe de ser excepcional. En niños se ha propuesto un posible rol de las MCP-I y II (monocyte chemotactic protein), promotoras de la síntesis de colágeno, en las enfermedades intersticiales pulmonares (99).
En el año 2003 se realizó una puesta al día de todo lo relacionado con el trasplante pulmonar pediátrico (100). Las enfermedades intersticiales pulmonares no se encuentran entre las causas más frecuentes del mismo. La fibrosis quística (41%) ocupa el primer lugar seguida de la hipertensión pulmonar primaria o secundaria (25%).
En resumen, el diagnóstico y clasificación de las neumonías intersticiales idiopáticas todavía son problemáticos para los patólogos. La reciente clasificación de consenso define los criterios clínicos, radiológicos y patológicos de cada una de las entidades (11). En caso de carecer de suficiente información clínico-radiológica puede ser útil, desde el punto de vista histológico, la clasificación según patrones morfológicos (23). Los nuevos avances en la TACAR, el refinamiento de los criterios patológicos y mayor número de datos de la historia natural de los procesos pueden ayudar a mejorar nuestros conocimientos sobre su evolución, patogenia y terapia (28,43). Por último, en algunos tipos de enfermedades pulmonares difusas infantiles hereditarias se han reconocido diversos tipos de alteraciones genéticas en las proteínas del surfactante (101,102). Estos hallazgos han impulsado el análisis de determinados polimorfismos genéticos de las proteínas del surfactante y del antagonista del receptor de IL-1, en estudios poblacionales amplios, que pueden dar luz sobre determinadas alteraciones constitucionales que predispongan a la fibrosis pulmonar idiopática (103,104).
AGRADECIMIENTOS
Mi gratitud más sincera a la Dra. Petra de la Torre y al Dr. Agustín Oñatibia por su crítica del manuscrito y las sugerencias aportadas.
BIBLIOGRAFÍA
Xaubet A. Consideraciones sobre la nueva clasificación de las neumopatías intersticiales difusas. Med Clin (Barc) 2003; 121: 389-95.
Raghu G, Brown KK. Interstitial lung disease: clinical evaluation and keys to an accurate diagnosis. Clin Chest Med 2004; 25: 409-19.
Steerle MP, Speer MC, Loyd JE, Broown KK, Heron A, Slifer SH, Burch LH, Wahidi MM et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med 2005; 172: 446-52.
Lee HL, Ryn JH, wittmer MH, Hartman TE, Lymp JF, Tazelaar HD, Limper AH. Familial idiopathic pulmonary fibrosis. Clinical features and outcome. Chest 2005; 127: 2034-41.
Allann JS, Limper AH. Idiopathic pulmonary fibrosis: is it a familial disease? Curr Opin Pulm Med 2006; 12: 312-7.
Leslie KO. Historical perspective: a pathologic approach to the classification of idiopathic interstitial pneumonias. Chest 2005; 128 (suppl): 513s-9s.
Noble PW, Homer RJ. Historical perspective on pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2005; 33: 113-20.
Liebow AA, Carrington DB. Potchen EJ, Le May M editors. Frontiers of pulmonary radiology. New York: Grune & Stratteon; 1969. p 102-41.
Kazenstein ALA, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157: 1301-15.
Katzenstein AL, Myers JL. Nonspecific interstitial pneumonia and the other idiopathic interstitial pneumonias: classification and diagnostic criteria. Am J Surg Pathol 2000; 24: 1.
American Thoracic Society/European Respiratory Society International multidisciplinary consensus classification of the idiopatic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277-304.
Zompatori M, Sverzellati N, Poletti V, Bnà C, Ormiti F, Spaggiari E, Maffei E. High-resolution CT in diagnosis of diffuse infiltrative lung disease. Sem Ultrasoud CT MRI 2005; 26: 332-47.
Quigley M, Hansell DM, Nicholson AG. Interstitial lung disease- the new synergy between radiology and pathology. Histopathology 2006; 19: 334-42.
Pipavath S, Godwin JD. Imaging of the chest: idiopathic interstitial pneumonia. Clin Chest Med 2004; 25: 651-6.
Brody AS. Imaging considerations: interstitial lung disease in children. Radiol Clin N Am 2005; 43:391-403.
Flaherty KR, King TE, Raghu G, Lynch JP, Colby V, Travis WD et al. Idiopatic interstitial pneumonia. What is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170: 904-10.
Wells AU. Histopathologic diagnosis in diffuse lung disease. An ailing gold standard. Am J Respir Crit Care Med 2004;170: 828-9.
King TE Jr. Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med 2005; 172: 268-79.
Nicholson AG, Addis BJ, Bharucha H et al. Inter-observer variation between pathologists in diffuse parenchymal lung disease. Thorax 2004; 59: 500-5.
Fishbein M. Diagnosis: To biopsy or not biopsy. Assessing role of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Chest 2005 (suppl) 128: 520s-5s.
Flaherty KR, Travis WD, Colby TV et al. Histopathologic variability in usual and nonsp3ecific interstitial pneumonias. Am J Respir Crit Care Med 2001; 164: 1722-7.
Riley DJ, Costanzo EJ. Surgical biopsy: its appropiateness in diagnosing interstitial lung disease. Curr Opin Pulm Med 2006; 12: 331-6.
Leslie KO. Pathology of interstitial lung disease. Clin Chest Med 2004; 25: 657-703.
Gaensler EA, Carrington CB. Open biopsy for chronic diffuse infiltrative lung disease: clinical, roentgegraphic and physiological correlation in 502 patients. Ann Thorac Surg 1980; 30: 411-26.
du Bois RM. Evolving concepts in the early and accurate diagnosis of idiopathic pulmonary fibrosis. Clin Chest Med 2006; 27: s17-s25.
Souza CA, Müller NL, Flint J, Wright JL, Churg A. Idiopathic pulmonary fibrosis: spectrum of high-resolution CT findings. AJR 2005; 81: 240-6.
Bjoraket JA, Ryn JH, Edwin MK et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 157: 199.
Travis WD, Colby TV, Koss MN, Rosado-de-Christenson ML, Müller NL, King TE Jr. Non-neoplastic disorders of the lower respiratory tract. Atlas of nontumor pathology. First series. Fascicle2. Washinton: AFIP; 2002. p. 49-114.
King Jr TE, Schwarz MJ, Brow K et al. Idiopathic pulmonary fibrosis. Relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164: 1025-32.
Myer JL, Katzenstein ALA. Fibroblasts on focus. Am J Respir Crit Care Med 2006; 174: 623-5.
King Jr TE, Tooze JA, Schwarz MI et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Resp Crit Care Med 2001; 164: 1171.
du Bois RM. Fibroblastic foci. Time to be counted? Chest 2006; 130: 3-4.
Noble PW. Idiopathic pulmonary fibrosis: natural history and prognosis. Clin Chest Med 2006; 27: 11s-6s.
Enomoto N, Suda T, Kato M, Kaida Y, Nakamura Y, Imokawa S, Ida M, Chida K. Quantitative análisis of fibroblastic foci in usual interstitial pneumonia. Chest 2006; 130: 22-9.
Cool CD, Groshong SD, Rai PR, Henson PM, Stewart S, Brown KK. Fibrobast foci are not discrete sites of lung injury or repair. The fibroblast reticulum. Am J Respir Crit Care Med 2006; 174: 654-8.
Ternamian PJ. Interstitial lung disease in myopathic dermatomyositis, dermatomyosistis and polimyositis. Eur Respir J 2003; 22: 245-50.
Tathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyosistis. Curr Opin Rheumatol 2005; 17: 701-6.
Highland KB, Silver RM. New developments in scleroderma interstitial lung disease. Curr Opin Rheumatol 2005; 17: 737-45.
King TC Jr. Nonspecific interstitial pneumonia and systemic sclerosis. Am J Respir Crit Care Med 2002; 165: 1578-9.
Martinez FJ. Idiopathic interstitial pneumonia. Proc Am Thorac Soc 2006; 3 81-95.
Witschi H, Haschek WM, Meyer KR, Ullrich RL, Dalbey WE. A pathogenic mechanism in lung fibrosis. Chest 1980; 78: 395-9.
Katzenstein AL. Pathogenesis of «fibrosis» in interstitial pneumonia: an electron microscopic study. Hum Pathol 1985; 16: 1015.
Noble PW, Home RJ. Idiopathic pulmonary fibrosis: new insights into pathogenesis. Clin Chest Med 2004; 25: 749-58.
Lappi-Blanco E, Kaarteenaho-Wiik R, Salo S, Sormunen R, Määkkö P. Laminin-5 gamma2 chain in cryptogenetic organizing pneumonia and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2004; 169: 27-33.
Tang YW, Johnson JE, Bronwing PJ et al Herpesvirus DNA is consistently detected in lug of patients with idiopathic pulmonary fibrosis. J Clin Microbiol 2003; 41: 2633.
Bradley TD, Millar YE, Martinez FJ, Angus DC, MacNec W, Abraham E. Interstitial lung disease, lung cancer, lung transplantation, pulmonary vascular disorders, and sleep-disordered breathing in AJRCCM in 2004. Am J Respir Crit Care Med 2005; 171: 675-85.
Zhang HY, Phan SH. Inhibition of myofibroblast apoptosis by transforming growth factor beta (1). Am J Respir Cell Mol Biol 1999; 21: 658.
Strieter RM. Pathogenesis and natural history of usual interstitial pneumonia: the whole story or the last chapter of a long novel. Chest 2005; 128 (suppl): 526s-32s.
Zuo F, Kaminski N, Eugeri E et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA 2002; 99: 6292-7.
Ebina M, Shimizukawa M, Shibata N, Kimura Y, Suzuki T et al. Heterogeneous increase in CD-34 positive alveolar capillaries in idipathic pulmonary fibrosis. Am J Respir Crit Care Med 2004; 170: 1203-8.
Cosgrove GP, Brown KK, SchiemannWP, Serls AE, Parr JE et al. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis. A role in aberrant angiogenesis. Am J Respir Crit Care Med 2004; 170: 242-51.
Chua F, Sly PD, Laurent GJ. Pediatric lung disease: from proteinases to interstitial pulmonary fibrosis. Pediatr Pulmonol 2005; 39: 392-401.
Odajima N, Betsuyaku T, Nasuhara Y, Itoh T, Fukuda Y, Senior RM, Noshimura M. Extracellular matrix metalloproteinase inducer interstitial pneumonia. Hum Pathol 2006; 37: 1058-63.
Ingrann JL, Bonner JC. EGF and PDGF receptor tyrosine kinases as therapeutic targets for chronic luna diseases. Curr Mol Med 2006; 6: 409-21.
Strieter RM, Belperio JA, Keane MP. Host innate defences in the lung: the role of cytokines. Curr Opin Infect Dis 2003; 16: 193.
Wilborn J, Crofford LJ, Burdick MD et al. Cultured lung fibroblast isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to repress cyclo-oxygenase-2. J Clin Invest 1995; 95: 1861.
Raghu G, Brown KK, Bradford WZ et al. A placebo-controlled trial of interferon (gamma)-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med 2004; 350: 125-33.
Strieter RM, Starko KM, Enelow RI, Noth I, Valentine VG et al. Effects of interferon-gamma 1b on biomarker expression in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2004; 170: 133-40.
Nathan S. Therapeutic intervention: assessing the role of the international consensus guidelines. Chest 2005; 128 (suppl): 533s-9s.
Raghu G, Brown KK, Bradford WZ et al. A placebo-controled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N England J Med 1999; 341: 1264-9.
Burdick MD, Murray LA, Keane MP et al. CXCL11 attenuates bleomycin-induced pulmonary fibrosis via inhibition of angiogenesis. Am J Respir Crit Care Med 2005; 171: 261-8.
Nathan SD. Therapeutic management of idiopathic pulmonary fibrosis: an evidence-based approach. Clin Chest Med 2006; 27: s25-s35.
Orens JB, Estenne M, Arcasoy S, Conte JV, Corris P, Egan JJ, Egan T, Keshavjee S et al. International guidelines for the selection of lung transplant candidates: 2006 update- A consensus report from the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplat 2006; 25: 745-55.
Wells AV, Desai SR, Rubens MB et al. A composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 2003; 167: 962-9.
www.ishlt.org/registries/slides.asp.
Grupo de Trabajo de la SEPAR. Trasplante pulmonar. Arch Bronconeumol 2001; 307-15.
Lynch DA, Godwin D, Safrin S, Starko KM, Hormel P et al. High-resolution computed tomography in idiopathic pulmonary fibrosis. Diagnosis and prognosis. Am J Respir Crit Care Med 2005; 172: 488-93.
McLoud TC. Role of high-resolution computed tomography in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2005; 172: 408-9.
Honma K, Abraham JL, Chiyotani K, de Vuyst P, Dumortier P, GibbvsAR, et al. Proposed criteria for mixed-dust pneumoconiosis: definition, descriptions, and guidelines for pathologic diagnosis and clinical correlation. Hum Pathol 2004; 35: 1515-23.
Acosta-Fernandez O. Marcadores séricos en la fibrosis pulmonar idiopática. Implicación pronóstica. Arch Bronconeumol 2006; 42: 377-9.
Gal AA, Staton GW. Current concepts in the classification of interstitial lung disease. Am J Clin Pathol 2005; 123 (suppl 1): 67-81.
Nagai S, Handa T, Tabuena R, Kitaichi M, Izumi T. Nonspecific interstitial pneumonia: a real clinical entity? Clin Chest Med 2004; 25: 705-15.
Hunninghake GW, Lynch DA, Galvin JR et al. Radiological findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest 2003; 124: 1215-25.
Martinez FJ, Keane MP. Update in diffuse parenchymal lung disease 2005. Am J Respir Crit Care Med 2006; 173: 1066-71.
MacDonald SLS, Rubens MB, Hausell DM. Non-specific interstitial pneumonia and usual interstitial pneumonia: comparative appearances at and diagnostic accuracy of thin section CT. Radiology 2001; 221: 600-5.
Tannickal VJ, Wells AV. Classification of interstitial pneumonias. What do gene expression profiles tell us? Am J Respir Crit Care Med 2006; 173: 141-2.
Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A. Gene expresión profiles distinguís idiopathic pulmonary fibrosis from hypersensivity pneumonitis. Am J Respir Crit Care Med 2006; 173: 188-98.
Travis WD, Matsui K, Moss J et al. Idiopathic non-specific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns; survival comparation with usual interstitial pneumonia and descamative interstitial pneumonia. Am J Surg Pathol 2000; 24: 29-33.
Myers JL, Veal CF, Shin MS, Katzenstein AL. Respiratory bronchiolitis causing interstitial lung disease: a clinicopathologic study of six cases. Am Rev Resp Dis 1987; 135: 880-4.
Davies G, Wells AU, du Bois RM. Respiratory bronchiolitis associated with interstitial lung disease and descamative interstitial pneumonia. Clin Chest Med 2004; 25: 717-26.
Vourlekis JS. Acute interstitial pneumonia. Clin Chest Med 2004; 25: 739-48.
Beasley MB, Franks TJ, Galvin JR, Travis WD. Acute fibrinous and organizing pneumonia. Arch Pathol Lab Med 2002; 126: 1064-70.
Cordier JF. Cryptogenetic organizing pneumonia. Clin Chest Med 2004; 25: 727-38.
Schelesinger C, Koss MN. The organizing pneumonias: an pdate and review. Curr Op Pulm Med 2005; 11: 422-30.
Ramírez Olivencia G, Martín Borge V, Sancho Bueso T. Bronquiolitis obliterante con neumonía organizada inducida por carbamazepina. Med Clin 2007; 128: 197-9.
Fan LL, Deterding RR, Langston C. Pediatric interstitial lung disease revisited. Pediatr Pulmonol 2004; 38: 369-78.
Shannon DC, Mark EJ. Celular interstitial pneumonitis in infants- a clinicopathological study. Chest 1992; 101: 1065-9.
Canakis AM, Cutz E, Manson D, O’Brodovich H. Pulmonary interstitial glucogenosis: a new variant of neonatal interstitial lung diseases. Am J Respir Crit Care Med 2002; 165: 1557-65.
Katzenstein AL, Gordon LP, Oliphant M, Swender PT. Chronic pneumonitis of infancy- a unique form of interstitial lung disease occurring in early childhood. Am J Surg Pathol 1995; 19: 439-47.
Hartl D, Griese M. Interstitial lung disease in children- genetic background and associated phenotypes. Respiratory Research 2005; 6: 32-48.
Hartl D, Griese M. Surfactant protein D in human lung disease. Eur J Clin Invest 2006; 36: 423-35.
Nogee LM. Genetics of pediatric interstitial lunf disease. Curr Opin Pediatr 2006; 18: 287-92.
Sherlenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fetal surfactant deficiency. N Engl J Med 2004; 350: 1296-303.
Bullard JE. ABCA3 mutations associated with pediatric interstitial lung diseases. Am J Crit Care Med 2005; 172: 1026-31.
Edwards V, Cutz E, Viero S, Moore AM, Nogee L. Ultrastructure of lamellar bodies in congenital surfactant deficiency. Ultrastr Pathol 2005; 29: 503-9.
Deterling R, Fan LL. Surfactant dysfunction mutations in children´s interstitial lung disease and beyond. Am J Respir Crit Care Med 2005; 172: 940-1.
Nicholson AG, Florio R, Hansell DM, du Bois RM, Wells AV, Hughes P, Ramadan HK, Mackinlay CI et al. Pulmonary involvement by Niemann-Pich disease. A report of six cases. Histopathology 2006; 48: 596-603.
Mueller GA, Pickoff AS. Pediatric lymphocytic interstitial pneumonitis in a HIV-negative child with pulmonary Epstein-Barr virus infection. Pediatr Pulmonol 2003; 36: 447-9.
Hartl D, Griese M, Nicolai T, Zissel G, Prell C, Reinhardt D et al. A role for MCP-I/CCR2 in interstitial lung disease in children. Respir Res 2005; 6: 93-104.
Sweet SC. Pediatric lung transplantation: update 2003. Pediatr Clin N Am 2003; 50: 1393-417.
Garcia CK. Inherited interstitial lung disease. Clin Chest Med 2004; 25: 421-33.
Raghu G, Mageto Y. Genetic predisposition of interstitial lung disease. En: Schwarz M, King T, editores. Interstitial lung disease. 3rd edition. Yearbook 1998. Hamilton (Ontario): Mosby; 1998. p. 119-32.
Selman M, Lin HM, Montano M et a. l. Surfactant protein A and B genetic variants predispose to idiopathic interstitial fibrosis. Hum Genet 2003; 113: 542-50.
Whyte M, Hubbard R, Meliconi R et al. Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor alpha gene polymorphisms. Am J Respir Crit Care Med 2000; 162 (2 Pt 1): 755-8.
![]()