
Vol. 41, n.º 4, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 4, 2008 |
REVISIONES
Manuel Vaquero
Hospital Donostia. Paseo Doctor Begiristain, 115.
20014 San Sebastián.
mvaquero@chdo.osakidetza.net
RESUMEN
El propósito de este artículo es discutir una aproximación práctica para pacientes con tumores del grupo del neuroblastoma. Se recomienda la utilización de la terminología, clasificación y de los criterios morfológicos del sistema pronóstico propuesto por el INPC (International Neuroblastoma Pathology Committee). Además, se recogen los datos necesarios que deben figurar en el informe anatomopatológico, incluidos los test citogenéticos y moleculares.
Palabras clave: Neuroblastoma, ganglioneuroblastoma, tumores del sistema nervioso simpático.
SUMMARY
The purpose of this article is to discuss a practical approximation for patients with tumours of the neuroblastic group. We recommend using the terminology, classification and morphological criteria of the prognostic system proposed by the INPC (International Neuroblastoma Pathology Committee). Furthermore, the necessary data for the anatomopathological report has been collected, including the cytogenic and molecular tests.
Keywords: Neuroblastoma, ganglioneuroblastoma, sympathetic nervous system tumours.
INTRODUCCIÓN
Los tumores del grupo de neuroblastomas periféricos son tumores embrionarios del sistema nervioso simpático. Es un grupo heterogéneo no sólo morfológicamente sino en su genética y biología. Hace más de dos décadas Shimada y col (1). sentaron las bases morfológicas para la clasificación de los tumores neuroblásticos que junto al estadio constituyen los factores pronósticos más importantes.
Los tumores neuroblásticos se caracterizan por una serie de hechos que los hacen únicos para el patólogo y el clínico. El patólogo debe manejar la pieza en fresco y prever la recogida de tejido para realizar estudios moleculares y citogenéticos. El clínico debe realizar una estadificación que no sigue la habitual TNM (tabla 1), así como analizar los resultados analíticos de metabolitos de catecolaminas en orina –ácido vanililmandélico y homovanílico–, y los niveles séricos de LDH, ferritina y enolasa neuroespefícica.

VARIABLES CLÍNICAS (1,2)
El 76% de los tumores se localiza en el abdomen, generalmente en las adrenales, seguido de la cadena simpática paravertebral y de paraganglios como el de Zuckerkandl. Los tumores adrenales afectan a los de menor edad y suelen ser más indiferenciados. El mediastino posterior ocupa el segundo lugar y pueden ser el origen de síntomas respiratorios y de síndrome de Horner. En lactantes la presentación puede ser una hepatomegalia debida a la presencia de metástasis difusas, o un síndrome de compresión radicular o medular, al extenderse el tumor radicularmente hasta el espacio epidural en forma de «reloj de arena». Estas ramificaciónes justifican que en casi un 70% de pacientes queden secuelas neurológicas posquirúrgicas de diverso tipo. Un síndrome paraneoplásico cerebeloso infrecuente es el opsoclono-mioclono que suele indicar buen pronóstico ya que se debe a la presencia de anticuerpos frente al tumor pero con reactividad cruzada con las células de Purkinje. Rara vez aparece una diarrea acuosa secundaria a la liberación de péptico vasointestinal por el tumor o un síndrome de Cushing. Hay casos familiares y otros asociados a síndromes como el de Beckwith-Wiedeman. También puede estar presente en el Hirschprung y en la neurofibromastosis, así como complicación del síndrome hidantoínico.
La prueba de imagen más rápida y útil en los de localización abdominal es la ecografía. La que más datos aporta es la tomografía axial computerizada de alta resolución (TACAR) con contraste, que informa no solo de la localización tumoral precisa sino también de su vascularización y extensión ganglionar. En la estadificación se utiliza el escáner óseo para descartar la afectación del sistema esquelético, lo que indicaría mal pronóstico.
En orina deben determinarse metabolitos de catecolaminas útiles tanto en el diagnóstico como en el seguimiento de los pacientes (2). La determinación del ácido vanililmandélico tiene una sensibilidad de 80% y una especificidad del 97%, mientras que el ácido homovanílico ofrece unas cifras de 72% y 98% respectivamente. En algunos casos, como en algunos neuroblastomas indiferenciados, pueden no estar elevados debido a la escasa actividad de la enzima dopamina- beta hidroxilasa.
A nivel sérico (2,3) suelen determinarse, antes de iniciar cualquier tratamiento, LDH, enolasa neuroespecífica (ENS) y ferritina. La LDH indicaría actividad de la enfermedad y valores por encima de 1.000 U/L suelen indicar mal pronóstico. Igual sucede con valores de ENS mayores de 30 ng/mL. Los valores de ferritina deben determinarse antes de realizar transfusiones sanguíneas y parece ser el mejor de los tres para indicar un peor pronóstico, cuando sobrepasa el valor de 142 ng/mL.
CATEGORÍAS MORFOLÓGICAS Y MANEJO DE LA MUESTRA
La biopsia debe recibirse inmediatamente después de su obtención, sin fijar y debe manejarse en condiciones que aseguren la mayor esterilidad posible. Es muy importante el reconocimiento de posibles nódulos macroscópicos (fig. 1). Para los estudios genéticos y moleculares son necesarios al menos 100 mg de tejido. Si hay material suficiente se hacen dos cubos de 1 cm de lado, de diferentes zonas, y se dividen en cuatro trozos (2):
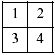

Fig. 1:
NB pobremente diferenciado. Las zonas
blanquecinas son calcificaciones del estroma. La abundancia de nidos separados
por estroma da un aspecto micronodular a la superficie de corte. Zonas de límite
abrupto, como la indicada con las flechas, deben estudiarse para descartar un GN
nodular.
1. Se hacen 10 improntas sin fijar y se fija el resto en formol para comparar con lo que se ha analizado.
2. Se introduce en medio de cultivo estéril, para citometría de ADN y citogenética.
3 y 4. Se congelan y mantienen en N2 líquido (–80ºC).
También puede tomarse material en fijador específico para estudio ultraestructural. Del resto de tumor debe hacerse un amplio muestreo, así como buscar la existencia de ganglios linfáticos. En España existe un centro de referencia para el estudio molecular del tumor (contacto con la secretaría del hospital infantil La Fe de Valencia: canyete_ade@gva.es / castel_vic@gva.es).
Microscópicamente siempre hay dos tipos celulares: neuroblastos y células de Schwann (1). Estas últimas serían reactivas, reclutadas por sustancias originadas en el propio tumor, y actuarían como antiproliferativas e inductoras de la maduración neuroblástica. Según la proporción de estos dos componentes hablamos de tumores ricos en estroma schwanniano, de buen pronóstico, y aquellos con menos del 50% de estroma en los que el pronóstico morfológico depende de otras variables como: la presencia de neuropilo y ocasionalmente de rosetas de Homer Wright, el porcentaje de maduración a células ganglionares y el índice mitótico-kariorréxico (MKI).
El MKI consiste en estimar el número de células en mitosis-kariorrexis en relación con el número total de células tumorales (2), ambos por campo de gran aumento (CGA). Debe cuantificarse en los tumores pobres en estroma. Es en esta categoría de neuroblastomas donde tiene una relevancia pronóstica. Conlleva los siguientes pasos:
• Valoración de la densidad celular (cortes de 3-4 µ):
– Intensa: 700 a 900 células /CGA.
– Moderada: 400 a 600 células /CGA.
– Escasa: 100 a 300 células /CGA.
– Mixta
• Elección al azar de los campos microscópicos para el recuento. En los de densidad intensa suelen ser suficientes 6 a 8 CGA mientras que en los escasos con mucho neuropilo debe recogerse al menos 20 CGA.
Se hace recuento de al menos 5.000 células neoplásicas, utilizando zonas diferentes del tumor y se anota la media del número de mitosis y núcleos en kariorrexis.
Las categorías que se obtienen son las siguientes (4):
MKI bajo: < de 100 cel MK/5.000 cel. tumorales o < 2% de células MK.
MKI intermedio: entre 100 y 200 cel MK/5.000 cel tumorales o entre el 2-4% de células MK.
MKI alto: >200 cel MK/5.000 cel tumorales o >4% células MK.
Hay correlación entre un MKI alto y un curso clínico adverso, así como con la amplificación de MYCN. Debe recordarse que el complejo proteico Myc-Max previene la maduración y promueve la proliferación y la apoptosis.
En la categoría histológica (2,4) de neuroblastomas hay, por definición, menos de del 50% de estroma schwanniano (figs. 2 y 3). En el subtipo indiferenciado no hay neuropilo ni diferenciación a células gangliónicas. El diagnóstico se hace con la ayuda de las técnicas especiales. Al igual que el subtipo pobremente diferenciado puede tener células grandes pleomórficas o de aspecto rabdoide.
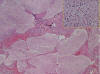
Fig. 2:
Aspecto microscópico del caso anterior con zonas
de calcificaciones del estroma (flecha) y abundante neuropilo pero sin células
ganglionares (recuadro).
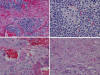
Fig. 3:
Distintos aspectos madurativos de los NB. A)
Abundante estroma junto a nidos neuroblásticos indiferenciados; B) roseta de
Homer-Wright; C) Presencia de células neuronales bien constituidas: amplio
citoplasma eosinófilo y nucleolos evidentes; y D) Zona con aspecto de
ganglioneuroma maduro.
Los neuroblastomas pobremente diferenciados tienen neuropilo en microscopía óptica. La mayoría de células tumorales son indiferenciadas y el número de células en diferenciación ganglionar no supera el 5%.
El neuroblastoma en diferenciación se diferencia del anterior por su abundante neuropilo pero el criterio base para clasificarlo es la presencia de más de un 5% de maduración a células ganglionares. Este subtipo puede tener abundantes células de Schwann en la periferia pero, a diferencia de los ganglioneuromas entremezclados, no supera el 50% del tumor. Ese cambio de celularidad periférico plantea la posibilidad de confusión con un tipo nodular pero, sin embargo, los bordes entre ambas zonas no son ni nítidos ni compresivos.
El ganglioneuroblastoma entremezclado es rico en estroma y con aspecto de diferenciación-maduración neuronal evidente. Sin embargo, el proceso de maduración no es completo ya que quedan focos microscópicos de células neuroblásticas (menos del 50% del volumen total de las laminillas). Debe recordarse que en los ganglioneuroblastomas nodulares los focos indiferenciados son macroscópicos.
En la categoría de ganglioneuromas hay dos subtipos, ambos ricos en estroma schwanniano y con células ganglionares maduras. En la forma «en maduración» todavía quedan neuroblastos en diferenciación o células ganglionares madurando pero no llegan a formar nidos. Por el contrario en la forma «madura» el proceso madurativo es completo y hay incluso fascículos de neuritas y células satélites perineuronales.
La categoría más compleja es la de ganglioneuroblastoma nodular (6,7) que contiene más de una clona evolutiva con diferente comportamiento biológico (tabla 2). El límite de los nódulos suele ser nítido e incluso con una pseudocápsula que separa al nódulo del resto. A veces se calcifican y pueden quedar ocultos. Deben estudiarse todos los nódulos y es el más indiferenciado el que, junto a la edad y el MKI, marca el pronóstico (tabla 3).

En el informe anatomopatológico de los tumores neuroblásticos deben figurar los siguientes datos:
Categoría histológica (tabla 2).
Subtipo histológico.
Presencia o no de invasión venosa y linfática.
MKI en la categoría de neuroblastoma (3).
Otros datos de interés: calcificación (podría indicar pronóstico favorable en relación al tratamiento), cantidad de neuropilo y de necrosis; así como si hay desgarros en la superficie por la cirugía (esto ocurre más frecuentemente si hay hemorragia tumoral difusa o degeneración quística).
Posible afectación de ganglios linfáticos y órganos adyacentes, para poder realizar la estadificación.
Resultados de los estudios especiales.
Determinación del grupo pronóstico según la INPC: histología favorable o desfavorable (tabla 3).
La información histológica obtenida en una metástasis es equivalente a la del sitio primario, aunque debe indicarse este hecho en el informe. Por el contrario, después de tratamientos quimio y/o radioterápicos la histología varía mucho de área a área y hay frecuentemente necrosis y calcificación por lo que no valdría para la valoración pronóstica. Sin embargo el MYCN sigue siendo útil.
Está documentado que los neuroblastomas pueden madurar y regresar, como en el estadio 4S. Así, todos los ganglioneuromas fueron una vez neuroblastomas y por eso se ven en las edades más tardías. La presencia en la adrenal de nidos neuroblásticos (2), en 1/220 recién nacidos y lactantes de menos de tres meses, no está aclarado si son restos embrionarios, hiperplasia o una verdadera neoplasia que luego regresaría.
El diagnóstico diferencial morfológico (3) se plantea fundamentalmente en los neuroblastomas indiferenciados o con muestras muy escasas o de mala calidad. Deben incluirse fundamentalmente: linfomas y leucemias, rabdomiosarcoma y PNET/Ewing. En estos casos la inmunohistoquímica (2,5) suele ser de gran ayuda (fig. 4) –NSE, cromogranina A, sinaptofisina– y debe recordarse que el anticuerpo CD 99, que detecta la glucoproteína de membrana p30/32 producto del gen MIC2, es negativo en los neuroblastomas. En algunos casos la proteina ALK puede resultar positiva (2). La microscopía electrónica también puede ayudar al permitir detectar gránulos citoplásmicos de tipo neurosecretor y/o neuritas con microtúbulos y terminaciones sinápticas (2).
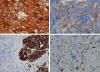
Fig. 4:
Inmunohistoquímica del NB: A) Tinción difusa con
enolasa neuroespecífica; B) Abundantes gránulos citoplásmicos con cromogranina
A; C) Patrón de tinción multifocal para neurofilamentos; y D) Presencia
incipiente de células de Schwann, S-100 positivas, en torno a nidos de
neuroblastos indiferenciados.
TEST CITOGENÉTICOS Y MOLECULARES (2,3)
La realización de estas pruebas necesita tejido fresco y las más utilizadas son aquellas implicadas en demostrar la amplificación de MYCN y la existencia de alteraciones cromosómicas.
El MYCN se localiza en el cromosoma 2 y para detectar su amplificación se está generalizando el uso de FISH, aunque también puede utilizarse el southern blot. Se amplifica en forma de regiones dobles diminutas que se tiñen homogéneamente. El resultado es que se produce una gran cantidad de proteína que forma complejos Myc-Max en el núcleo de la célula tumoral, lo que inhibe su diferenciación y promueve la proliferación y apoptosis. Suele resultar amplificado en los neuroblastomas indiferenciados y en los pobremente diferenciados con MKI alto.
La amplificación de MYCN se correlaciona, a nivel de citogenética, con la delección 1p en estadios avanzados, en los que también aparecen otras delecciones desfavorables como la 14q, 11q y la ganancia en 17q. La LOH del 1p precede al desarrollo de la amplificación MYCN.
El índice de ADN detectado mediante citometría de flujo también tiene relación con el pronóstico, sobre todo en pacientes menores de un año. Mientras la hiperdiploidía/casi triploide se asocia a buen pronóstico, ocurre lo contrario con los casi diploides/tetraploides.
Por último, la expresión alta de TrkA –un factor de crecimiento neural de alta afinidad– sobre todo presente en los neuroblastomas de histología favorable, se asocia a buen pronóstico, mientras que la glucoproteína de superficie CD44 se correlaciona con la amplificación MYCN.
PRONÓSTICO
Después de ajustar el estadio y la histología, favorable o no, el pronóstico es semejante entre los nodulares y aquellos que no lo son. Los estadios 1 sólo con cirugía alcanzan casi el 100% de supervivencia libre de recaída (4,8). Mientras los 2A y 2B, sin amplificación de MYCN, alcanzan el 98% de supervivencia sin enfermedad a los cinco años si tienen categoría favorable y un 73% si la tienen desfavorable. Por el contrario en los estadios 4 la supervivencia baja al 3%, mientras en el grupo especial de 4S varía entre el 60 y el 90%. Las metástasis más frecuentemente afectan al hígado y a los huesos (cráneo y órbita). La mayoría de las recidivas se producen en los dos primeros años tras la escisión (2).
BIBLIOGRAFÍA
Shimada H, Chatten J, Newton WA, Sachs N, Hamondi AB, Chiba T, Marsden HB, Misugi K. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. JNCI 1984; 73: 405-16.
Rosai and Ackerman´s Surgical Pathology. J Rosai. 9 th Ed. Mosby 2004. Vol. 1; Cap. 16; pag. 1127-35.
Qualman SJ, Bowen J, Fitzgibbous PL, Cohn SL, ShimAda H for the members of the Cancer Committre, College of American Pathologists. Protocol for the examination os specimens from the patioens with neuroblastoma and related neuroblastic tumors. Arch Pathol Lab Med 2005; 129: 874-83.
Navarro S, Amann G, Beiske K, Cullimanee CJ, d´amore ESG, Gambini C, et al. Prognostic value of international neuroblastoma pathology classification in localized respectable peripheral neuroblastic tumors: A histopathologic study of localized neuroblastoma European Study Group 94.01 trial and protocol. J clin Oncol 2006; 24: 695-9.
Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald. Terminology and morphologic criteria of neuroblastic tumors. Recommendations by the International Neuroblastoma Pathology Committee. Cancer 1999; 86: 349-63.
Penchmaur M, d’Amore ESG, Joshi VV, Hata J, Roald B, Dehener LP, et al. Revision of the International Neuroblastoma Pathology Classification. Confirmation of favourable and unfavourable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 2003; 98: 2274-8.
Umehara S, Makagawa A, Lukens JN, Seeger RC, Stram DD, Gerbing RB, Shimada H. Histopathology defines prognostic subsets of ganglioneuroblastoma, nodular. A reprint from the Children’s Cancer Group. Cancer 2000; 89: 1150-61.
Burgues O, Navarro S, Noguera R, Pellín A, Ruiz A, Castel V, Llombart-Bosch A. Prognostic value of the International Neuroblastoma Pathology Classification in Neuroblastoma (Schwannian stroma-poor) and comparison with other prognostic factors: a study of 182 cases from the Spanish Neuroblastoma Registry. Virchows Archiv 2006; 449: 410-20.
![]()