
Vol. 42, n.º 1, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 1, 2009 |
CASUÍSTICA
Margarita Gimeno Aránguez1, Enrique de Álava Casado2, Emilio Álvarez Fernández1
1 Servicio de
Anatomía Patológica. Hospital General Universitario Gergorio Marañón. Madrid.
2 Centro de Investigación del Cáncer IBMCC. Salamanca.
mgimeno.hgugm@salud.madrid.org
RESUMEN
El liposarcoma mixoide aparece en extremidades de pacientes jóvenes, y muestra una apariencia mixoide, y unas características citogenéticas determinantes.
Presentamos el caso de un varón de 37 años con un gran tumor retroperitoneal, que había sido diagnosticado previamente de un tumor de muslo. El tumor se caracterizaba por presentar una red vascular capilar plexiforme con células fusiformes monomorfas y células redondas atípicas. Mostraba una translocación (12;16). Planteó el diagnostico diferencial con liposarcoma desdiferenciado con áreas de tipo mixofibrosarcoma.
El liposarcoma mixoide en retroperitoneo es más probable de origen secundario, por la peculiar propensión del LPS mixoide de extremidad a dar metástasis en tejidos blandos extrapulmonares, sobre todo retroperitoneo.
Palabras clave: Liposarcoma, mixoide, metástasis, retroperitoneo.
SUMMARY
Myxoid liposarcoma usually ocurs in extremities in younger patients, showing a myxoid appearance and distinctive cytogenetic features.
We report a huge retroperitoneal myxoid tumor, with in a patient that initially presented a liposarcoma of the thigh. The tumour showed a fine plexiform capillary network with monomorphic spindle-cell and atypical round cells. FISH showed a translocation t(12;16). This retroperitoneal myxoid tumor may be confused with dedifferentiated liposarcoma with myxofibrosarcoma-like areas.
The retroperitoneal myxoid liposarcoma is more likely to be secondary. Retroperitoneum is the most common site of extrapulmonary metastatic myxoid liposarcoma.
Keywords: Liposarcoma, myxoid, metastases, retroperitoneal.
INTRODUCCIÓN
Los sarcomas de retroperitoneo constituyen un grupo amplio de tumores primarios de malignidad variable, y representan un 10% de todos los sarcomas (1). Los más frecuentes son los liposarcomas (61%), seguidos de los leiomiosarcomas, fibrohistiocitomas malignos y sarcomas sinoviales.
Los liposarcomas se clasifican en: bien diferenciado, mixoide y de células redondas, desdiferenciados y pleomórficos. En retroperitoneo son más frecuentes los LPS bien diferenciados y desdiferenciados (2). Los LPS mixoides y de células redondas son un subtipo histológico diferente de los anteriores que ocurre predominantemente en extremidades (3-5).
Presentamos el caso de un varón joven que desarrolla una tumoración de gran tamaño en retroperitoneo, con la característica translocacion t(12;16) de los LPS mixoides, que se trataba de la metástasis de un LPS mixoide primario en muslo. Revisamos la literatura y planteamos el diagnóstico diferencial con otros sarcomas mixoides, la evolución peculiar de los LPS mixoides, y la necesidad de realizar técnicas de biología molecular para el diagnóstico de sarcomas mixoides.
CASO CLÍNICO
Se trata de un varón de 37 años que presenta una tumoración intramuscular de muslo izquierdo 14 x 12 x 10 cm, superficie de corte fasciculada blanco grisácea con áreas de aspecto gelatinoso brillante mixoide.
Histológicamente se trata de una neoplasia mesenquimal maligna con patrón vascular capilar plexiforme, abundante celularidad monomorfa de células estrelladas de pequeño tamaño en el seno de estroma mixoide. En otras zonas se evidencian lagos mixoides centrado por vasos de pequeño calibre, con concentración celular perivascular (fig. 1).
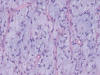
Fig. 1:
Áreas de liposarcoma mixoide típico con celulas
monomorfas estrelladas separadas por abundante estroma mixoide y red vascular
plexiforme.
Muestra áreas con mayor densidad celular con células de mayor tamaño, núcleos con discreto nucleolo y cromatina abierta, que constituyen un 10% de la neoplasia, con escaso estroma mixoide intercelular (fig. 2). Índice de proliferación celular Ki 67 30% en las áreas de mayor densidad celular (fig. 3).
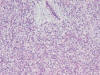
Fig. 2:
Áreas de liposarcoma de mayor densidad celular
con células redondas y menor cantidad de estroma mixoide entre ellas.
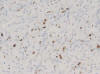
Fig. 3:
Técnica IHQ para la expresión de Ki 67 30%.
Se realizó tratamiento con resección quirúrgica amplia y bordes libres.
A los tres años el paciente presenta una tumoración retroperitoneal derecha de 30 x 15 cm que engloba el riñón.
En el estudio histológico de esta neoplasia se observa una proliferación mesenquimal densamente celular de células redondas con escaso estroma mixoide entre ellas, núcleos con nucleolo fino y cromatina abierta. Las células se encuentran densamente empaquetadas, dejando ver con dificultad una red vascular plexiforme capilar entre ellas, que se hace más evidente con técnicas inmunohistoquímicas CD 34+. También se observan lipoblastos, de citoplasma multivacuolado y núcleo central, con improntas en el contorno nuclear. Estas áreas constituyen el 60% de la neoplasia (fig. 4). Se observan áreas de liposarcoma clásico con estroma mixoide, zonas de lagos mixoides hipocelulares y perivasculares (fig. 5). El índice de proliferación celular Ki 67 es de 50% en las zonas de mayor densidad celular (fig. 6).
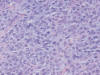
Fig. 4:
Tumor retroperitoneal densamente celular, de
células redondas separadas por escaso fondo mixoide, que dificulta la
visibilidad de red vascular plexiforme.
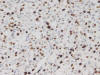
Fig. 5:
Expresión IHQ de Ki 67 50%.
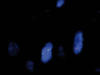
Fig. 6:
FISH utilizando las sondas FUS Breakapart de
Vysis. Se trata de un cóctel de sondas que marcan cada extremo del gen con un
fluorocromo rojo y verde, respectivamente. Se observan imágenes de separación en
los núcleos celulares evaluables en al menos un 30% de los núcleos.
En ambas tumoraciones de muslo y retroperitoneo, se realizan técnicas de FISH para la demostración de translocación 12;16 fusión (FUS-DDIT3 ), que se observa en ambos casos (fig. 7).
A los 9 meses el paciente vuelve a presentar una recidiva intraabdominal, que engloba tronco arterial celíaco, lo que le ocasiona la muerte.
DISCUSIÓN
El LPS mixoide y de células redondas constituye un 30 a 35% de todos los LPS (3), reconociéndose en la última clasificación de la OMS (5 ) como un subtipo único, que presenta una translocación especifica t(12;16) que se produce por la fusión del gen DDIT3 (CHOP) con el gen FUS (TLS) , y con características clínicas que le hacen diferente de los otros LPS : en varones jóvenes, miembros inferiores y con una peculiar propensión a dar metástasis extrapulmonares (2-5).
La localización del LPS mixoide y de células redondas en retroperitoneo es excepcional y cuando se presentan deben diferenciarse de otros sarcomas mixoides, de liposarcomas desdiferenciados con componente mixoide, y metástasis, como ocurre en nuestro caso (2,5).
Los LPS mixoides son considerados de bajo grado, pero se han revisado los criterios morfológicos de estos tumores, siendo la hipercelularidad un criterio de mal pronóstico y agresividad, con mayor propensión a las recurrencias y metástasis (6). Como hemos visto en nuestro caso, el tumor primario tiene áreas hipercelulares que constituían un 10% de la tumoración, progresando hasta un 60% en la metástasis retroperitoneal. Como demuestra Smith (6 ) en su trabajo sobre LPS mixoides y de células redondas de extremidades, los pacientes con un porcentaje superior al 5% de componente de células redondas en el tumor inicial tenían una proporción alta de metástasis o muerte. La transición entre estos tumores no está clara y se debería a anomalías cromosómicas secundarias, siendo el grado histológico en relación a la hipercelularidad el factor pronóstico más importante de los LPS mixoides (3,7).
En los trabajos de Pearlstone (8) y Estourgie (9), demuestran una alta tendencia de los LPS mixoides de dar metástasis extrapulmonares , superior comparativamente a otros LPS, y el sitio más frecuente era retroperitoneo, seguido de mesenterio, pared abdominal anterior, tejidos blandos de espalda, con una clara disminución de la supervivencia media que descendía en enfermedad localizada del 73% al 57% al presentar metástasis.
Estourgie (9) observa que los LPS mixoides tienen alta predilección por las metástasis extrapulmonares, hasta tres veces superior a las pulmonares (29%), signo de mal pronóstico en la evolución de estos pacientes y porque se asocia a una disminución de la supervivencia, que es de 12 meses después de la metástasis. Por lo tanto, los pacientes con LPS mixoide deben ser evaluados en su seguimiento con técnicas radiológicas para un diagnostico temprano de metástasis en retroperitoneo, e intrabdominales, ya que suponen un claro empeoramiento de su evolución clínica.
El tratamiento quirúrgico de lesiones iniciales puede evitar complicaciones más severas, sin implicación de otros órganos intraabdominales y retroperitoneales.
El LPS mixoide muestran una translocación especifica t(12;16)(q13;p11), y la generación de un gen de fusión FUS-DDIT3 (TLS-CHOP), en el 90% de los LPS mixoides, o la variante t(12;22) en que el gen DDIT3 (CHOP) se fusiona con EWS en 2% de los LPS mixoides. Ambas fusiones suponen un marcador genético altamente específico que les diferencia de otros tumores mixoides (7,10,11).
Hay lesiones mixoides «simuladoras», con las que se debe hacer el diagnóstico diferencial, y deben utilizarse técnicas genéticas moleculares para distinguir el LPS mixoide fundamentalmente de mixofibrosarcomas, y LPS desdiferenciados con áreas mixoides, LPS bien diferenciados mixoides, condrosarcoma mixoide extraesquelético, y lesiones benignas como lipomas con cambios mixoides, mixoma y schwannomas mixoides (3,7,11,13).
En retroperitoneo nos plantea el diagnóstico diferencial fundamentalmente con los LPS desdiferenciados, que se caracterizan morfológicamente por presentar junto a áreas de LPS bien diferenciado Lipoma-Like o esclerosante, otras zonas mixoides con abundantes celulas pleomorficas y red vascular capilar prominente, en arcadas, similar a los mixofibrosarcomas.. Estos hallazgos morfológicos son importantes y claves para diferenciarlos de los LPS mixoides, con red vascular plexiforme bien organizada, y celularidad monótona fusiforme, o redonda.
La tendencia a dar metástasis en partes blandas fue considerada como multicentricidad, con tumores sincrónicos o metacrónicos, pero la realización de ciertos estudios genéticos demostró el origen monoclonal de estos tumores multifocales, estableciendo la naturaleza metastásica de las lesiones en tejidos blandos distales (12).
Por lo tanto deben aplicarse estas pruebas moleculares en el diagnóstico de tumores mixoides (11,13). Actualmente la hibridación in situ con fluorescencia (FISH) con sondas DNA específicas en improntas o material fijado en parafina ha supuesto un método útil para el diagnóstico de tumores sólidos, especialmente en el caso de translocaciones cromosómicas específicas, mediante sondas de fusión o de ruptura. Se ha demostrado su validez para el diagnostico y se puede utilizar en material de archivo (13).
Los sarcomas de retroperitoneo muestran en común el gran tamaño que suelen alcanzar y la dificultad en todos ellos de una resección quirúrgica completa, dos criterios fundamentales para el pronóstico de estos tumores. El tipo histológico, el grado y la expresión de marcadores oncogénicos como P53 son fundamentales para predecir la evolución de los LPS mixoides y por lo tanto para su manejo inicial, seguimiento e indicación de otros tratamientos (14).
El tratamiento del LPS mixoide es quirúrgico con resecciones amplias y bordes libres, y la radioterapia intraoperatoria supone gran ayuda para el manejo de estos tumores, siendo aún de mayor ayuda en los localizados en retroperitoneo, donde la resección quirúrgica es de gran dificultad.
Por lo tanto, ante el hallazgo de liposarcoma mixoide en retroperitoneo, se debe descartar que se trate de la metástasis de un primario en extremidad, y utilizar técnicas citogenéticas moleculares para el diagnóstico de su translocación específica.
BIBLIOGRAFÍA
Bautista N, Wendy S, O’Connell T. Retroperitoneal soft-tissue sarcomas: Prognosis and treatment and recurrent disease. Am Surg 2000; 66: 832-6.
Enzinger FM, Weiss SW: Liposarcoma (chapter 17) Soft Tissue Tumors , 4rd ed. Edited by Enzinger FM, Weiss SW, St Louis MO Mosby 2001, pg. 641-95.
Orvieto E, Furlanetto A, Laurino L, Dei Tos AP. Myxoid and round cell liposarcoma: a spectrum of myxoid adipocitic neoplasia Semin Diagn Pathol 2001; 18: 238-49.
Weiss S. Lipomatous tumors. In soft tissue tumors Monographs in Pathology Edited by Weiss S, Brooks J. Georgia. Williams and Wilkins 1996, 207-39.
Antonescu C, Ladanyi M. Myxoid liposarcoma. In Pathology and genetics of tumours of soft tissue and bone Editores Fletcher CDM, Unni K.K. Mertens F. WHO Lyon 2002, pg. 40-3.
Smith TA, Easley KA, Goldblum JR: Myxoid/Round cell liposarcoma of the extremities. A clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma. Am J Surg Pathol 1996; 20: 171-80.
Meis-Kindblom JM, Sjogren H, Kindblom LG, Peydro-Mellquist A. Cytogenetic and molecular genetic analyses of liposarcoma and its soft tissue simulators: recognition of new variants and differentiala diagnosis. Virchows Arch 2001; 439: 141-51.
Pearlstone DB, Pisters PWT, Bold RJ, Feig BW, Hunt KK, Yasko AW, Patel S, Pollack A, Benjamin RS, Pollock RE. Patterns of recurrence in extremity liposarcoma. Implications for staging and follow-up. Cancer 1999; 85: 85-92.
Estourgie S, Nielsen GP, Ott MJ. Metastatic patterns of extremity myxoid liposarcoma and their outcome. J Surg Oncol 2002; 80: 89-93.
Tallini G, Akerman M, Dal Cin P, de Wever I, et al. Combined morphologic and karyotypic study of 28 myxoid liposarcomas : implications for a revised morphologic typing (a report from the CHAMP group) Am J Surg Pathol 1996; 20: 1047-55.
Antonescu CR, Elahi A, Humphrey M, Lui MY, Healey JH, Brennan MF, Woodruff JM, Jhanwar SC, Ladanyi M. Specificity of TLS-CHOP rearrangements for classic myxoid/Round cell Liposarcoma. Absence in predominantly myxoid well-differentiated liposarcoma J Mol Diag 2000; 2: 132-8.
Antonescu CR, Elahi A, Healey JH, Brennan MF, Lui MY, Lewis JJ, Jhanwar SC, Woodruff JM, Ladanyi M. Monoclonality of multifocal myxoid liposarcoma confirmation by analysis of TLS-CHOP or EWS-CHOP rearrangements. Clin Cancer Res 2000; 6: 2788-93.
Aoki T, Hisaoka M, Kouho H, Hashimoto H, Nakata H. Interphase cytogenetic analysisi of myxoid soft tissue tumors by fluorescence in situ hybridisation and DNA flow cytometry using paraffin-embebed tissue Cancer 1997; 79: 284-93.
Antonescu CR, Tschernyavsky SJ, Decuseara R, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res 2001; 7: 3977-87.
![]()