
Vol. 42, n.º 2, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 2, 2009 |
CASUÍSTICA
José Ferrando Marco1, M.ª Jesús Arilla Morell2, José Tomás Navarro3
1 Servicio de
Anatomía Patológica. Hospital Arnau de Vilanova.
2 Servicio de Hematología. Hospital de Sagunto.
3 Servicio de Hematología. Hospital Germans Trías y Pujol de
Badalona.
ferrando_josmar@gva.es
RESUMEN
Paciente varón de 19 años, con sospecha clínica de Enfermedad de Crohn, intervenido quirúrgicamente por un cuadro clínico de oclusión intestinal del segmento distal del yeyuno. El estudio de la pieza quirúrgica mostró una tumoración que infiltraba la pared intestinal y grasa mesentérica adyacente. Histológicamente se evidenció una infiltración de la pared intestinal por células poco diferenciadas que demostraron positividad a la mieloperoxidasa, lo que confirmó el diagnóstico de sarcoma mieloide. No se encontró evidencia de afectación de médula ósea, enfermedad leucémica ni síndrome mielodisplásico o mieloproliferativo. Se instauró tratamiento quimioterápico y tras una serie de complicaciones que se resolvieron favorablemente, el paciente permanece asintomático a los 24 meses del diagnóstico inicial. El interés del caso estriba no sólo en la rareza de este tipo de tumores y la ubicación intestinal en un paciente sin enfermedad hematológica previa, sino también en la dificultad del diagnóstico anatomopatológico, dado que un 75% de estos casos aleucémicos son erróneamente diagnosticados de inicio y confundidos con otros tumores malignos pobremente diferenciados.
Palabras clave: Sarcoma granulocítico, sarcoma mieloide, cloroma, tumor mieloide extramedular.
SUMMARY
A 19 year-old man with a clinical suspicion of Crohn’s disease underwent surgery for intestinal occlusion in the distal jejunum. Macroscopically, a mass infiltrating the intestinal wall and adjacent mesenteric fat was seen. Histologically, the intestinal wall was infiltrated by poorly differentiated cells which were positive for myeloperoxidase and a diagnosis of myeloid sarcoma was made. No evidence of bone marrow involvement or myelodysplastic or myeloproliferative disorders was seen. The patient was treated with chemotherapy and, after several complications which were successfully resolved, is asymptomatic 2 years after the initial diagnosis. This unusual case of a rare tumour, occurring in the intestine in the absence of previous haematological malignancy, draws attention to the diagnostic difficulties involved; indeed, 75% of nonleukemic cases are initially misdiagnosed, often being confused with poorly differentiated malignant tumours.
Keywords: Granulocytic sarcoma, myeloid sarcoma, chloroma, extramedular myeloid tumor.
INTRODUCCIÓN
El sarcoma mieloide (SM) es una masa tumoral localizada compuesta por células inmaduras de la serie granulocítica mieloide (1,2). Puede presentarse como complicación en pacientes con leucemia mieloide aguda, leucemia mieloide crónica, síndromes mielodisplásicos o mieloproliferativos y esporádicamente en sujetos asintomáticos (1-4). Las localizaciones extramedulares mas habituales son ganglios linfáticos, hueso, piel y partes blandas, siendo menos frecuente la localización gastrointestinal (6,5%) (4-7).
La importancia de conocer este tipo de tumores estriba en el hecho de que un 75% de los casos sin enfermedad hematológica son erróneamente diagnosticados de inicio (1,3,8,9). Un 25% de estos tumores evolucionan durante meses o años, sin progresar al estado leucémico, incluso se han descrito casos de curación completa, generalmente tras radioterapia local y/o quimioterapia sistémica (2-4,10).
Aportamos un caso de SM de localización yeyunal en un paciente de 19 años con cuadros suboclusivos durante año y medio que hicieron pensar en una enfermedad de Crohn.
CASO CLÍNICO
Varón de 19 años que ingresa con un cuadro clinico de oclusión intestinal. El paciente, sin antecedentes familiares de interés, estaba en vigilancia clínica desde hacía año y medio por sospecha de enfermedad de Crohn yeyunoileal, en tratamiento con Azatioprina y Omeprazol y pendiente de ser llamado para intervención quirúrgica. En el exámen físico se apreciaba delgadez y palidez cutánea. A la exploración abdominal se palpaba una masa dura y desplazable en hipogastrio compatible con asa o plastrón inflamatorio. En el estudio radiológico previo al cuadro de ingreso se realizó ecografía, TAC abdominal y tránsito intestinal que informaron de una masa adenopática que englobaba pared intestinal y provocaba estenosis yeyunal y dilatación de asas preestenóticas. La gammagrafía con leucocitos marcados no evidenció actividad inflamatoria. La analítica mostró normalidad en todos sus parámetros tanto hematimétricos cómo bioquímicos. Se indicó la intervención quirúrgica realizándose una laparotomía media y visualizándose una masa yuxtayeyunal de unos 6 centímetros que infiltraba la pared intestinal y provocaba estenosis. Se resecó dicha masa junto al segmento yeyunal realizándose anastómosis término-terminal. La biopsia intraoperatoria fue informada de malignidad como linfoma, por lo que no se actuó sobre otra masa adenopática próxima a vasos yeyunales que descansaban sobre la mesentérica superior. El postoperatorio evolucionó con normalidad.
Estudio anatomopatológico: La pieza quirúrgica mostraba un segmento de intestino delgado de 9 cm con engrosamiento del tercio medio. La disección mostró una masa tumoral de 4 cm de aspecto gris claro, homogénea y de consistencia media que infiltraba la pared intestinal y protuía en la mucosa sin ulcerarla (fig. 1). Se incluyeron muestras en parafina y se realizaron secciones de 4-6 micras parte de las cuales fueron desparafinadas, rehidratadas y teñidas con con hematoxilina-eosina. A otras secciones se les practicó estudio inmunohistoquímico mediante el Sistema de visualización «EnVision» de Dako Corporation. Se utilizaron los anticuerpos primarios para mieloperoxidasa (prediluído comercial), CD117 (c-kit) (dilución 1:50), CD34 (dilución 1:50), CD68 (KP1, prediluído comercial), CD20 (L26, prediluído comercial) , CD3 (F7.2.38, dilución 1:50), citoqueratina (AE1/AE3, prediluído comercial), PS100 (dilución 1:50) y Melanosoma (HMB45, prediluído comercial), todos ellos así mismo de Dako Corporation.

Fig. 1:
Sección longitudinal de la pieza quirúrgica dónde
se aprecia la tumoración infiltrando la pared muscular y protuyendo en la mucosa
intestinal.
En el estudio histológico definitivo se apreciaba una infiltración de la pared intestinal (fig. 2) por células atípicas con escaso citoplasma y gruesos núcleos a menudo hendidos, con pequeños nucleolos y notable actividad mitótica (fig. 3). En mucha menor proporción se observaban células granulares eosinófilas de núcleos lobulados. En base a estos hallazgos se realizó el diagnóstico de tumor mieloide extramedular pobremente diferenciado o SM blástico . En el estudio inmunohistoquímico, las células neoplásicas mostraron positividad para mieloperoxidasa (fig. 4), CD117(c-kit) y CD34, lo que confirmó el diagnóstico. El estudio con sondas FISH sobre material parafinado descartó la presencia de t(8;21).
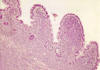
Fig. 2:
Aspecto microscópico panorámico que muestra la
infiltración de la mucosa intestinal. H-E x100.
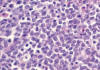
Fig. 3:
Detalle de la celularidad neoplásica. Obsérvese
la lobulación y los finos límites nucleares, así cómo la presencia de
promielocitos dispersos. H-E x600.
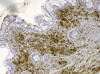
Fig. 4:
Positividad para mieloperoxidasa en las células
neoplásicas que infiltran la pared intestinal. PAP x200.
Tras el postoperatorio inmediato, el estudio hematológico que incluyó biopsia de médula ósea, no evidenció indicios de enfermedad leucémica, y la TAC mostró una masa abdominopélvica de 7 x 3 cm y pequeñas imágenes ganglionares de 0,5 cm en grasa mesentérica de íleon terminal.
Se incluyó al paciente en el protocolo quimioterápico CETLAM para menores de 70 años con profilaxis antibiótica y antifúngica. La TAC abdominopélvica posterior evidenció remisión completa de las adenopatías residuales. Durante el ingreso en el tratamiento de consolidación, presentó un proceso infeccioso que afectó bazo, pancitopenia, hipocaliemia severa y síntomas depresivos. El proceso infeccioso requirió finalmente esplenectomía, que se realizó sin complicaciones con buena evolución posterior, lo que a su vez facilitó la resolución de la pancitopenia. La hipocaliemia se resolvió bien con aporte de potasio y los síntomas depresivos mejoraron con ayuda psicológica y soporte de antidepresivos y ansiolíticos.
A 24 meses del diagnóstico, el paciente permanece asintomático de su proceso original y controlado en consulta de oncología y hematología.
DISCUSIÓN
El SM ha recibido diversas denominaciones; cloroma, sarcoma granulocítico y tumor mieloide extramedular (1,4,11). Warthin en 1904 fue el primero en asociar los SM a la leucemia mieloide aguda (11). La aparición de este tipo de tumores en sujetos asintomáticos, sin enfermedad leucémica ni medular, es poco frecuente (4,9,12). La incidencia es significativamente mayor en hombres (60-80%) que en mujeres, predominando en pacientes jóvenes con una media de edad entre 43 y 47 años (2-5,12). Las localizaciones extramedulares mas frecuentes son ganglios linfáticos, hueso, piel y partes blandas (2-4,7,8). La ubicación gastrointestinal representa un 6,5% de los casos en las grandes series publicadas (2,3,5) de los que la localización en intestino delgado representa un 11% (4). En la literatura anglosajona, encontramos reseñados 21 casos de SM de intestino delgado, de ellos 12 aleucémicos (4,6,8,10,13,14). Tal cómo ocurrió en nuestro caso, la manifestación clínica, generalmente en forma de dolor abdominal, distensión y vómitos, de duración variable, induce a diagnósticos erróneos cómo enfermedad de Crohn, apendicitis aguda u oclusión intestinal de origen indeterminado, que llevan a la intervención quirúrgica (6,7,11,14).
El diagnóstico de este tipo de tumores es anatomopatológico. Clásicamente se han distinguido tres categorías de diferenciación en los SM: blásticos, inmaduros y bien diferenciados (2-4,10). Los bien diferenciados (21%), contienen variable proporción de neutrófilos y eosinófilos maduros así cómo de promielocitos, siendo escasa la cantidad de mieloblastos. Estos últimos abundan más en los inmaduros (32%). Los blásticos, entre los que se encuentra nuestro caso, son los más frecuentes (47%), y en ellos la celularidad predominante son mieloblastos (1,2,9). Las formas inmaduras y las blásticas, en especial estas últimas, son las que presentan mayor problema de diagnóstico diferencial. En las series publicadas de casos aleucémicos, el 75% los diagnósticos fueron erróneos, 50% lo fueron de linfomas y el 25% restante de otros tumores indiferenciados. (2-4,15). El diagnóstico diferencial se plantea fundamentalmente con linfomas de alto grado, carcinomas pobremente diferenciados, melanomas, sarcoma de Ewing, rabdomiosarcoma y meduloblastoma (1,4,5,8,14). Histológicamente, en el SM existen tres hallazgos fundamentales de importancia diagnóstica. Por una parte un patrón infiltrativo peculiar, que respeta la arquitectura tisular, al contrario de lo que ocurre con los linfomas cuya tendencia es destructiva (14). En segundo lugar, las células blásticas contienen unos núcleos grandes, frecuentemente hendidos o seudolobulados y que muestran una delgada membrana nuclear, cromatina poco densa y pequeños nucleolos, mientras que en los linfomas la membrana y el nucleolo suelen ser gruesos y la cromatina densa. (1,4,14). Por último, en las formas inmaduras y diferenciadas, la presencia de promielocitos, mielocitos eosinófilos y neutrófilos maduros debe ser suficiente para al menos, en principio, pensar en esta entidad (1,2,14,15).
El diagnóstico definitivo hay que buscarlo en la tipificación de los marcadores que caracterizan a los elementos progenitores de la serie mieloide granulocítica, fundamentalmente la mieloperoxidasa, CD34 y el CD117 (c-kit) (2-5,8,11,13,16). En una serie de 30 casos a los que se aplicó un amplio panel de trece anticuerpos, la mieloperoxidasa con un 97% y el CD117(c-kit) con un 87% de positividad, fueron los anticuerpos mas idóneos para confirmar el diagnóstico de este tipo de tumores (16). Ambos anticuerpos resultaron positivos en nuestro caso.
No está muy claro de qué forma el tratamiento puede influir en la supervivencia en los casos sin enfermedad hematológica. Muchos pacientes evolucionan a LMA en plazos que oscilan entre 10 y 44 meses (2,6,12), con una supervivencia media de 22 meses (6,12). El tratamiento quirúrgico, coadyuvado con quimioterapia cómo si se tratase una LMA, es la pauta mayoritariamente utilizada en las series revisadas, acompañadas de radioterapia local si se trata de una tumoración unifocal (2,3,4,8,12). Con esta pauta, se pueden alcanzar supervivencias en algunos casos de hasta ocho años e incluso remisón completa (3,4,8,12).
BIBLIOGRAFÍA
Brunning RD. Leukemias and related disorders. Myeloid Sarcoma. En: Rosai J, editor. Rosai and Ackerman’s Sugical Pathology. 9.ª edición. Philadelphia: Mosby; 2004; 2064-6 .
Neiman RS, Barcos M, Berard C, Bonner H, Mann R, Rydell RE et al. Granulocytic Sarcoma. A clinicopathologic study of 61 biopsied cases. Cancer 1981; 48: 1426-37.
Meis JM; Butler JJ, Osborne BM, Manning JT. Granulocytic Sarcoma in nonleucemic patients. Cancer 1986; 58: 2697-709.
Kohl SK, Aoun P. Granulocytic Sarcoma of the Small Intestine. Arch Pathol Lab Med 2006; 130: 1570-4.
Menasce LP, Banerjee SS, Beckett E, Harris M. Extramedullary myeloid tumour (granulocytic sarcoma) is often misdiagnosed: A study of 26 cases. Histopathology 1999; 34: 391-8.
Corpechot Ch, Lémann M, Brocheriou I, Mariette X, Daniel Mth, Berteau Ph et al Granulocytic Sarcoma of the jejunum: A rare case of small bowel obstruction. Am.J. of Gastroenterology 1998; 93: 2586-8.
Beck ThM, Day JC, Smith ChE, Hugh E. Eddy. Granulocytic Sarcoma treated as a acute leukemia. Report of a case. Cancer 1984; 53: 1764-6.
Paydas S, Zorludemir S, Ergin M. Granulocytic Sarcoma: 32 cases and review of the literature. Leukemia and Lymphoma 2006; 47: 2527-41.
Brunning RD, Matutes E, Flandrin G, Vardima J, Bennett J, Head D et al. Acute Myeloid Leukemia not otherwise categorized. Myeloid Sarcoma. Tumours of haematopoietic and lymphoid tissues. WHO Classification of tumours. Lyon. IARC Press: p. 104-5.
Zhou W, Vasquez JC, O’Donnell MR, Paz BI. Clinical manifestations of gastrointestinal granulocytic sarcoma requiring surgical treatment. Am Surg 2007; 67(8): 764-6.
Brugo EA, Larkin E, Molina-Escobar J, Constanzi J. Primary granulocytic sarcoma of the small bowell. Cancer 1975; 35: 1333-40.
Imrie KR, Kovacks MJ, Selby D, Lipton J, Patterson BJ, Pantalony D et al. Isolated Chloroma: The effect of early antileukemic therapy. Ann Intern Med 1995; 123(5): 351-3.
Palomino-Portilla EA, Valbuena JR, Quiñónez-Avila MP, Medeiros LJ. Myeloid Sarcoma of appendix mimicking acute apendicitis. Arch Pathol Lab Med 2005; 129: 1027-31.
Mrad K, Abid L, Driss M, Ben Abid H, Ben Romdhane K. Granulocytic Sarcoma of the Small Intestine in a Child Without Leukemia. Acta Cytol 2004; 48: 641-4.
Suh Yae Kyung, Hyung Ju C. Shin. Fine-Needle aspiration biopsy of granulocytic sarcoma. A clinicopathologic study of 27 cases. Cancer 2000; 90: 364-72.
Chen J, Yanuck RR 3rd, Abbondanzo SL, Chu WS, Aguilera NS. c-Kit (CD117) Reactivity in Extramedullary Myeloid Tumor/Granulocytic Sarcoma. Arch Pathol Lab Med 2001; 125: 1448-51.
![]()