
Vol. 42, n.º 3, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 3, 2009 |
CASUÍSTICA
Belén Ferri-Ñíguez1, M.ª Amparo Torroba-Carón1, Joaquín Sola-Pérez1, Claudio Piqueras-Pérez2
Hospital Universitario «Virgen de la Arrixaca».
Ctra. Madrid-Cartagena, s/n. 30120 El Palmar. Murcia. España.
1 Servicio de Anatomía Patológica.
2 Servicio de Neurocirugía.
belenferri@msn.com
RESUMEN
El glioma cordoide del tercer ventrículo es un tumor cerebral poco frecuente, de reciente descripción, localizado en la región hipotalámica - III ventrículo. Hasta la fecha, tan sólo hay 56 casos publicados desde que se describiera por primera vez en 1998 por Brat y cols. Histológicamente, el tumor está compuesto por células poligonales dispuestas en nidos o cordones, inmersas en una matriz mucoide con un intenso infiltrado inflamatorio linfoplasmocitario. Las células neoplásicas coexpresan característicamente marcadores inmunohistoquímicos de Proteína Ácida Gliofibrilar (PAGF), Citoqueratinas, Vimentina y CD-34. A nivel ultraestructural muestra características de diferenciación ependimaria y recientemente también se han identificado alteraciones genéticas características distintas a las de los gliomas. La extirpación tumoral fue incompleta tras un abordaje anterior. Nosotros describimos un nuevo caso de este tumor en un varón de 26 años y revisamos la literatura.
Palabras clave: Glioma cordoide, ependimoma, tercer ventrículo.
SUMMARY
Chordoid glioma of the third ventricle is an infrecuent brain tumor, located the third ventricle-hypothalamic region. Described for the first time by Brat in 1998, only 56 cases have been described in the literature. Histologically, the tumor consisted of polygonal epithelioid cells embedded in a mucinous matrix with a prominent lymphoplasmacytic infiltrate. The tumor cells expressed Glial Fibrillary Acidic Protein (GFAP), Cytokeratins, Vimentin and CD-34. This neoplasm exhibits features of ependymal differentiation on ultrastructural examination. Another authors also suggest a distinct genetic origin from other gliomas with specific molecular characterization. An incomplete resection was performed via an anterior approach. A case in a 26 years old man is reported and a review of the literature is included.
Keywords: Chordoid glioma, ependymoma, third ventricle.
INTRODUCCIÓN
El Glioma cordoide del III ventrículo fue descrito por primera vez en 1998 por Brat y colaboradores (6) como una nueva entidad tumoral y fue reconocida por primera vez por la OMS en el año 2000 (4). Aquellos autores describieron una serie de 8 pacientes (7 mujeres y 1 varón) con edades comprendidas entre los 30 y 71 años, que presentaban una tumoración en el tercer ventrículo, de apariencia cordoide a nivel histológico, pero con un perfil inmunohistoquímico y ultraestructural peculiar. Estaban constituídos por cordones y nidos de células poligonales inmersas en una matriz basófila rica en mucinas y se acompañaban de un infiltrado inflamatorio linfoplasmocitario maduro. Inmunohistoquímicamente, se observó expresión de Proteína Acida Gliofibrilar (PAGF), Vimentina y positividad débil o negatividad para EMA. La histogénesis del tumor permanece desconocida pero muchos autores han propuesto un probable origen en la célula ependimaria por hallazgos a nivel ultraestructural de microvellosidades apicales, estructuras hemidesmosoma-like y formación focal de membrana basal y otros autores han propuesto un origen tanicítico (11,29,23,3).
Recientemente Horbinski y cols. (16) han detectado por FISH alteraciones genéticas en 9p21 y 11q13 en cinco gliomas cordoides.
Actualmente la OMS (clasificación 2007) (5) los incluye en el apartado de «Otros Tumores Neuroepiteliales» y los considera como un glioma grado II de malignidad. A pesar de su bajo potencial maligno, es importante considerar la elevada morbi-mortalidad postquirúrgica y las elevadas tasas de extirpación incompleta descritas en este tipo de tumores (27,41).
Presentamos un nuevo caso de glioma cordoide del III ventrículo en un varón de 26 años y revisamos la literatura publicada hasta la fecha.
CASO CLINICO
Varón de 26 años, sin antecedentes clínicos, que ingresa en nuestro hospital para estudio de un síndrome hipotalámico crónico con hipersomnia e hiperfagia. El paciente había engordado unos 40 kgrs. en dos años. Además, relataba una disminución progresiva de la agudeza visual del ojo derecho hasta convertirse prácticamente en una ceguera completa de dicho ojo.
En la Resonancia Magnética (RM) realizada en secuencias sagital T1W y coronal T2W y axial T2 FLAIR, se observó una tumoración en región supraselar, de localización extraaxial, de 3 x 2,8 x 4 cm (AP x T x L) (fig. 1). Producía compresión de la hipófisis, sin llegar a infiltrarla, e impronta y desplazamiento hacia arriba del hipotálamo produciendo un colapso de la pared anterior del III ventrículo hasta el agujero de Monro, sin evidencia de hidrocefalia. La lesión presentaba hiposeñal en T1 e hiperseñal en T2, con una región baja de señal a nivel central. Además existían zonas microquísticas múltiples en el centro de la lesión. El estudio se completó con contraste paramagnético observándose captación de contraste de predominio periférico con un área central no captante. Existía edema perilesional en la cara interna de ambos lóbulos temporales e hipotálamo.
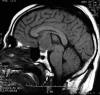
Fig. 1:
Imagen del tumor en la RM en un corte sagital en
la que se aprecia una tumoración sólida de bordes bien definidos, de
localización supraselar, que ocupa la porción anterior del III ventrículo.
Obsérvese la isointensidad del tumor respecto al parénquima cerebral adyacente.
Se realizó una craneotomía con un abordaje del III ventrículo vía transcallosa para la resección tumoral. Macroscópicamente la extirpación fue incompleta y los escasos fragmentos resecados fueron enviados al servicio de Anatomía Patológica. Microscópicamente se evidenció una proliferación neoplásica de células poligonales dispuestas en nidos y cordones, de citoplasmas amplios y eosinófilos, con núcleos redondos u ovoideos con cromatina fina sin atipias. En las áreas menos celulares e incluso a nivel intercelular, se observaba un estroma mucinoso y/o mixoide y microvacuolado. Además, llamaba la atención la existencia de agregados de células inflamatorias mononucleares linfoplasmocitarias tanto a nivel intratumoral como intersticial con ocasionales cuerpos de Russell (fig. 2). Fue difícil encontrar alguna figura de mitosis y el índice de proliferación celular Ki-67 resultó positivo en el 2% de las células tumorales. No se identificaron focos de necrosis, ni calcificaciones ni áreas tumorales con morfología de meningioma menongotelial clásico.
![]()
Fig. 2:
Detalles histológicos del glioma cordoide. A:
Area muy celular constituída por nidos y cordones de células poligonales de
citoplasmas eosinófilos en el seno de una matriz vacuolada (H-E 200x). B: Area
menos celular en la que predomina el estroma mucoide/mixoide con cordones e
hileras de células tumorales con infiltrado inflamatorio linfoplasmocitario
añadido (H-E 200x).
Inmunohistoquímicamente las células mostraron positividad intensa y difusa para PAGF, CD-34 y Vimentina, y parcheada o focal para CK AE1-AE3, EMA y proteína S-100. La matriz mucoide-mixoide mostró positividad con Azul Alcián (fig. 3).
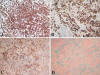
Fig. 3:
Inmunohistoquímica: A: Positividad intensa y
difusa de las células neoplásicas para PAGF (100x) (A) y CD-34 (200x) (B) y
parcheada y más débil con CK AE1-AE3 (200x) (C). D: Positividad del estroma con
Azul Alcián (200x).
Con estos resultados obtenidos el diagnóstico fue de glioma cordoide.
La Tomografía Axial Computerizada (TAC) de control realizada a las 12 horas de la intervención reveló una masa en la región selar, con extensión supraselar, de 2,5 x 2 x 3,4 cm (AP x T x L), que captaba contraste de forma homogénea y en anillo, compatible con persistencia tumoral. El paciente tuvo un cuadro confusional en el postoperatorio con desorientación temporo-espacial que progresivamente fue desapareciendo.
Transcurridos 5 meses desde la intervención quirúrgica, el paciente ha disminuído de peso y presenta, prácticamente, los mismos defectos visuales que antes de la cirugía. Sigue tratamiento con Topamax® para disminuir la clínica del síndrome hipotalámico.
DISCUSIÓN
El término de Glioma Cordoide fue acuñado por Brat y cols.(6) en 1998 para describir un tumor con características histológicas e inmunofenotípicas especiales, con una localización típica: la región anterior del III ventrículo. Posteriormente, fue en el año 2000, cuando la OMS (4) lo reconoció como una nueva entidad tumoral y lo clasificó en el apartado de «Tumores Neuroepiteliales de Origen Incierto» junto con el Astroblastoma y la Gliomatosis Cerebri. En la nueva clasificación de la OMS del año 2007 (5), se incluye en el capítulo de «Otros Tumores Neuroepiteliales» junto con el Astroblastoma y el Glioma Angiocéntrico. Son tumores raros, de crecimiento lento y no invasivos, carentes de necrosis tumoral, proliferación vascular y/o atipias, por lo que se consideran tumores de bajo grado de malignidad y se les asigna un grado II de la OMS.
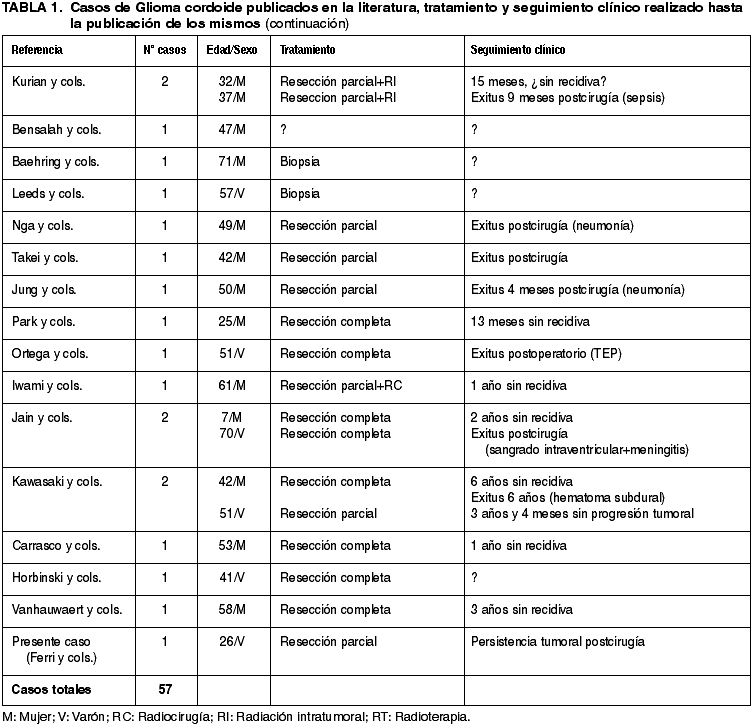
Tan sólo han sido publicados 57 casos de glioma cordoide en una década (incluyendo el presente) (tabla 1), siendo la mayor serie publicada de 8 pacientes en el trabajo original de Brat y cols.(6). De todos los casos contabilizados, se desprende que existe un ligero predominio en mujeres de la 5ª década (36 mujeres frente a 21 varones), con una media de edad de ambos sexos de 46.03 años, si bien han sido descrito dos casos en niños de 7 y 12 años (9,18) y otros dos casos en ancianos de 70 y 71 años respectivamente (18,1). En 1995, Wanschitz y cols. (42) publicaron un caso de un meningioma supraselar en una mujer de 24 años que expresó, curiosamente, positividad para PAGF en todas las células neoplásicas. Este caso representó, probablemente, el primer ejemplo de glioma cordoide publicado en la literatura y así ha sido recogido por la mayoría de autores en sus trabajos.
Los pacientes presentan gran variedad de síntomas clínicos derivados de su localización específica en la región anterior del III ventrículo y de la afectación de estructuras vecinas como el hipotálamo, la hipófisis, estructuras del quiasma óptico y lóbulos temporales. También pueden debutar con síntomas secundarios a una hidrocefalia obstructiva (cefaleas, naúseas). Son típicas las alteraciones endocrinas (hipotiroidismo, diabetes insípida, amenorrea), los defectos visuales y las alteraciones del comportamiento (alteración de la memoria, hipersomnia, hiperfagia, psicosis, insomnio) (27,38). Sólo en el caso del niño de 7 años (18) la localización fue yuxtaventricular y no la típica en la región anterior del III ventrículo. Otro de los casos fue un hallazgo casual descubierto en una TAC a una paciente tras sufrir un accidente de tráfico (7) (probablemente es el mismo caso que el publicado por Gallina y cols. (12) recientemente ya que coinciden todos los datos clínicos).
Radiológicamente se trata de un tumor bien circunscrito, generalmente sólido, que raramente puede presentar áreas de necrosis, calcificación (sólo en 2 de los casos) (17,19) o quistificación central (14,18,29,30,39). Suelen ser hiperdensos en las imágenes de TAC y en las imágenes de RM pueden ser isointensos en T1 e isointensos o ligeramente hiperintensos en T2, hallazgos que coinciden con los de nuestro caso. Además, es posible observar edema vasogénico bilateral por el efecto masa del tumor e hidrocefalia dependiendo de la localización del tumor (30). Muestran buena demarcación con la hipófisis y pueden invadir el hipotálamo y desplazar el infundíbulo posteriormente, hallazgos que los distingue, respectivamente, de los adenomas de hipófisis y de los hamartomas del tuber cinereum. Otra prueba de imagen que puede ser útil en el diagnóstico diferencial preoperatorio puede ser la RMN con perfusión. Los niveles obtenidos son significativamente más bajos que los del meningioma y los gliomas de alto grado (14).
Las características histopatológicas de este tumor incluyen la morfología poligonal de las células neoplásicas, con amplios citoplasmas eosinófilos, que se disponen en nidos y cordones en un estroma microvacuolado rico en mucinas que le aporta una apariencia «cordoide». Además, es característico que asocien un llamativo infiltrado inflamatorio de tipo linfoplasmocitario con ocasionales cuerpos de Russell. En todos los casos publicados están ausentes los rasgos de malignidad como es la atipia citológica, figuras de mitosis, necrosis o la proliferación vascular. El índice de proliferación celular suele ser muy bajo con cifras que oscilan entre 0 y 5% (6,21,24,8,32). En uno de los casos publicados, se identificaron algunos histiocitos y células gigantes multinucleadas tipo Touton en la periferia del tumor (33). Otros hallazgos adicionales inusuales descritos han sido la metaplasia condroide en un niño de 12 años (9) y la formación de papilas en el caso de una mujer de 57 años (31). Suh y cols. (36) describen un caso de glioma cordoide asociado a un quiste de Rathke y plantean que pueda corresponder a un tumor de colisión.
Los gliomas cordoides generalmente expresan positividad difusa para PAGF, Vimentina y CD-34 y expresión variable y focal de EMA, Citoqueratinas y proteína S-100 (6,7,11, 29,33). Aunque lo más común es encontrar negatividad para marcadores neuronales como Sinaptofisina, Neurofilamentos (NF) y Enolasa Neuronal Específica (ENE), algunos autores han identificado expresión de NF y de ENE y sugieren un origen divergente (neuronal y glial) de la neoplasia (7,17). La matriz mucinosa suele ser positiva para PAS y Azul Alcián.
El diagnóstico diferencial incluye otros tumores sólidos que surgen en la región del III ventrículo (astrocitoma pilocítico hipotalámico, ependimoma, craneofaringioma, adenoma de hipófisis, metástasis) aunque, por su apariencia «cordoide», el diagnóstico se establece fundamentalmente entre dos entidades: el meningioma cordoide y el cordoma. La ausencia de áreas tumorales de meningioma clásico y la ausencia de una intensa expresión de EMA, en contraste con la fuerte expresión de PAGF ayudan a excluirlo. Además, suele mostrar relación con duramadre y está asociado al Síndrome de Castleman (35). Por otro lado, el cordoma suele tener relación con hueso y está constituído fundamentalmente por células fisalíforas que coexpresan de forma intensa Citoqueratinas y proteína S-100 y suelen mostrar negatividad para PAGF.
La histogénesis del tumor permanece hoy en día confusa. A nivel ultraestructural se han identificado filamentos intermedios citoplasmáticos, microvellosidades apicales, estructuras hemidesmosoma-like y lámina basal focal. Por estos hallazgos, algunos autores defienden la hipótesis de un posible origen de la célula tumoral en las células ependimarias y un putativo origen en el órgano subcomisural (órgano secretor situado en la región dorsocaudal del III ventrículo desarrollado en el período fetal y que sufre involución postnatal) (11,29,38). Otros, sin embargo, apoyan que el glioma cordoide pudiera tener su origen en los tanicitos: células ependimarias especializadas, abundantes en la pared anterior del III ventrículo y completamente ausentes en el hipotálamo dorsal, con características citológicas intermedias entre los ependimocitos y astrocitos (31,34). Otros autores, sin embargo, apoyan un origen del glioma cordoide en células ependimarias especializadas de la lámina terminalis (región anterior del III ventrículo) basándose en la detección de vesículas de secreción y un patrón de zonación citológica y en estudios de neuroimagen (23, 3).
De los 57 casos de glioma cordoide publicados, tan sólo en 9 de ellos se han realizado estudios moleculares, quizás debido a la rareza del tumor. En 1999, Reifenberger y cols. (32), publicaron un trabajo en el que analizaban, por hibridación genómica comparada (CGH), 4 gliomas cordoides. A diferencia de meningiomas, gliomas y cordomas, no identificaron ni anomalías cromosómicas ni tampoco alteraciones en TP53 ni CDKN2A. Tampoco se identificaron amplificaciones de los proto-oncogenes EGFR, CDK4 o MDM2. Recientemente, Horbinski y cols. (16), han estudiado 5 casos más y, en consonancia con lo publicado previamente, tampoco identificaron mutaciones en TP53. Con técnica de FISH pudieron comprobar alteraciones genéticas diferentes a las observadas en gliomas convencionales como son las alteraciones en 9p21 y 11q13.
La resección tumoral fue completa o casi completa en 30 pacientes (52,63%) y parcial en 23 (40,35%) de ellos siendo algunos de los pacientes tratados con radiocirugía o radioterapia de forma adicional (8 pacientes; 14,03%). No se constató recidiva tumoral en ninguno de los pacientes que fueron sometidos a una resección completa, por lo que se deduce, que es el tratamiento de elección en este tipo de tumores. El papel de la radioterapia o radiocirugía está todavía por determinar debido a la escasez de pacientes tratados (19,22,25,41).
Del seguimiento clínico de los pacientes observamos que, las secuelas y complicaciones postquirúrgicas derivadas son graves y el pronóstico de los pacientes malo, en comparación con el bajo grado de malignidad de este tipo de neoplasias. Esto es debido fundamentalmente a la mala localización, con afectación de estructuras anatómicas regionales (hipotálamo), y al consiguiente difícil abordaje quirúrgico. De los 57 casos publicados, disponemos de seguimiento clínico en 45 de ellos (78,94%). Doce de los pacientes (22,64%) fallecieron en el postoperatorio inmediato, siendo el tromboembolismo pulmonar una de las causas más frecuentes (5/12). Otras causas fueron neumonía, traqueobronquitis, meningitis bacteriana e infarto agudo de miocardio (13,17,22,26).
CONCLUSIÓN
Los gliomas cordoides son tumores infrecuentes con una localicación, histología, perfil inmunohistoquímico y hallazgos moleculares peculiares. La resección tumoral completa parece ser el tratamiento de elección aunque en muchos de los casos no es viable por la localización e invasión de estructuras vecinas como el hipotálamo. Es necesario tener especial atención a estos pacientes en el seguimiento postoperatorio debido al alto riesgo de fenómenos tromboembólicos descritos.
BIBLIOGRAFÍA
Baehring JM, Bannykh S. Chordoid glioma of the third ventricle. J Neurooncol 2006; 76: 269.
Bensalah M, Cattin F, Katranji H, Viennet G, Bonneville JF. Case 6. Chordoid glioma. J Radiol 2005; 86: 531-2.
Brat DJ. The elusive origin of chordoid glioma. Arch Pathol Lab Med 2006; 130: 437-8.
Brat DJ, Scheithauer BW, Cortez SC, Reifenberger G, Burger PC. Chordoid glioma of the third ventricle. In: Kleihues P, Cavenee WK, editors. Pathology and genetics of tumours of the nervous system. 2nd ed. Lyon: IARC Press 2000; pp. 90-1.
Brat DJ, Scheithauer BW. Chordoid glioma of the third ventricle. In: David N. Louis, Hiroko Ohgaki, Otmar D. Wiestler, Webster K. Cavenee, editors. Who Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC Press 2007; pp. 90-1.
Brat DJ, Scheithauer BW, Staugatis SM, Cortez SC, Brecher K, Burger PC. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol 1998; 57: 283-90.
Buccoliero AM, Caldarella A, Gallina P, Di Lorenzo N, Taddei A, Taddei GL. Chordoid Glioma: clinicopathologic profile and differential diagnosis of an uncommon tumor. Arch Pathol Lab Med 2004; 128: e141-e145.
Carrasco R, Pascual JM, Reina T, Nieto S, Linera J, Sola RG. Chordoid Glioma of the third ventricle attached to the optic chiasm. Successful removal through a trans-lamina terminalis approach. Clin Neurol Neurosurg 2008; 110: 828-33.
Castellano-Sánchez AA, Schemankewitz E, Mazewski C, Brat DJ. Pediatric chordoid glioma with chondroid metaplasia. Pediatr Dev Pathol 2001; 4: 564-7.
Castellano-Sánchez AA, Recine MA, Restrepo R, Howard LH, Robinson MJ. Chordoid glioma: a novel tumor of the third ventricle. Ann Diagn Pathol 2000; 4: 373-8.
Cenacchi G, Roncaroli F, Cerasoli S, Ficarra G, Merli GA, Giangaspero F. Chordoid glioma of the third ventricle: an ultrastructural study of three cases with a histogenetic hypothesis. Am J Surg Pathol 2001; 25: 401-5.
Gallina P, Pansini G, Mouchaty H, Mura R, Buccoliero AM, Lorenzo ND. An incidentally detected third ventricle chordoid glioma. Neurol India 2007; 55(4): 406-7.
Galloway M, Afshar F, Geddes JF. Chordoid glioma: an uncommon tumour of the third ventricle. Br J Neurosurg 2001; 15: 147-50.
Grand S, Pasquier B, Gay E, Kremer S, Remy C, Le Bas JF. Chordoid glioma of the third ventricle: CT and MRI, including perfusion data. Neuroradiology 2002; 44: 842-6.
Hanbali F, Fuller GN, Leeds NE, Sawaya R. Choroid plexus cyst and chordoid glioma. Report of two cases. Neurosurg Focus 2001; 10: E5.
Horbinski C, Dacic S, McLendon RE, Cieply K, Datto M, Brat DJ, Chu CT. Chordoid glioma. A case report and molecular characterization of five cases. Brain pathol 2008; Jul 23 [Epub ahead of print].
Iwami KI, Arima T, Oooka F, Fukumoto M, Takagi T, Takayasu M. Chordoid glioma with calcification and neurofilament expression: case report and review of the literature. Surg Neurol 2008 Feb 8 [Epub ahead of print].
Jain D, Sharma MC, Sarkar C, Suri V, Rishi A., Garg A, Vaishya S. Chordoid glioma: report of two rare examples with unusual features. Acta Neurochir 2008; 150: 295-300.
Jung T-Y, Jung S. Third ventricular chordoid glioma with unusual aggressive behavior. Neurol Med Chir (Tokyo) 2006; 46: 605-8.
Kawasaki K, Kohno M, Inenaga C, Sato A, Hondo H, Miwa A, Fujii Y, Takahashi H. Chordoid glioma of the third ventricle: A report of two cases, one with ultrastructural findings. Neuropathology 2008 May 21 [Epub ahead of print].
Koh EJ, Choi HY. Chordoid glioma in the third ventricle. J Korean Neurosurg Soc 2003; 33: 501-4.
Kurian KM, Summers DM, Statham PFX, Smith C, Bell JE, Ironside JW. Third ventricular chordoid glioma: clinicopathological study of two cases with evidence for a poor clinical outcome despite low grade histological features. Neuropathol Appl Neurobiol 2005; 31: 354-61.
Leeds NE, Frederick FL, Ribalta T, Sawaya R, Fuller GN. Origin of chordoid glioma of the third ventricle. Arch Pathol Lab Med 2006; 130: 460-4.
Nakajima M, Nakasu S, Hatsuda N, Takeichi Y, Watanabe K, Matsuda M. Third ventricular chordoid glioma:case report and review of the literature. Surg Neurol 2003; 59: 424-8.
Nga ME, Tan KB, Laporte JP, Takano A. Test and teach. A recurrent third ventricular brain tumour. Diagnosis: Chordoid glioma of the third ventricle. Pathology 2006; 38: 254-7.
Oda M, Sasajima T, Kinouchi H, Sageshima M, Mizoi K. Third ventricular chordoid glioma: report of a surgical case. No Shinkei Geka 2002; 30: 973-9.
Ortega-Martínez M, Cabezudo JM, Bernal-García LM, Fernández-Portales I, Gómez-Perals L, Gómez de Tejada R, Pimentel JJ. Glioma cordoide del III ventrículo. Nuevo caso y revisión de la literatura. Neurocirugía (Astur) 2007; 18: 115-22.
Park SH, Hwang J-H. Chordoid glioma: an uncommon tumor of the third ventricle. J Korean Neurosurg Soc 2006; 40: 40-3.
Pasquier B, Péoc’h M, Morrison AL, Pasquier D, Grand S, Sindou M, Kopp N. Chordoid Glioma of the Third Ventricle. A report of two new cases, with further evidence supporting an ependymal differentiation, and review of te literature. Am J Surg Pathol 2002; 26: 1330-42.
Pomper MG, Passe TJ, Burger PC, Scheithauer BW, Brat DJ. Chordoid glioma: a neoplasm unique to the hypothalamus and anterior third ventricle. AJNR Am J Neuroradiol 2001; 22: 464-9.
Raizer JJ, Shetty T, Gutin PH, Obbens EA, Holodny AI, Antonescu CR, Rosenblum MK. Chordoid glioma: report of a case with unusual histologic features, ultrasestructural study and review of the literature. J Neurooncol 2003; 63: 39-47.
Reifenberger G, Weber T, Weber RG, Wolter M, Brandis A, Kuchelmeister K, Pilz P, Reusche E, Lichter P, Wiestler OD. Chordoid glioma of the third ventricle: immunohistochemical and molecular genetic characterization of a novel tumor entity. Brain Pathol 1999; 9: 617-26.
Ricoy JR, Lobato RD, Baez B, Cabello A, Martínez MA, Rodríguez G. Suprasellar chordoid glioma. Acta Neuropathol (Berl) 2000; 99: 699-703.
Sato K, Kubota T, Ishida M,Yoshida K, Takeuchi H, Handa Y. Immunohistochemical and ultrastructural study of chordoid glioma of the third ventricle: its tanycytic differentiation. Acta Neuropathol 2003; 16: 176-80.
Serrano Pardo R, Acevedo Barberá A. Meningioma cordoide. A propósito de un caso y revisión de la literatura. Rev Esp Patol 2007; 40: 243-6.
Suh YL, Kim NR, Kim JH, Park SH. Suprasellar chordoid glioma combined with Rathke´s cleft cyst. Pathol Int 2003; 53: 780-5.
Takei H, Bhattacharjee MB, Adesina AM. Chordoid glioma of the third ventricle: Report of a case with cytologic features and utility during intraoperative consultation. Acta Cytol 2006; 50: 691-6.
Taraszewska A, Bogucki J, Andrychowski J, Kozewski W, Czernicki Z. Clinicopathological and ultrastructural study in two cases of chordoid glioma. Folia Neuropathol 2003; 41:175-82.
Tonami H, Kamehiro M, Oguchi M, Higashi K, Yamamoto I, Njima T, Okamoto K, Akai T, Iizuka H. Chordoid glioma of the third ventricle: CT and MR findings. J Comput Assist Tomogr 2000; 24: 336-8.
Vajtai I, Varga Z, Scheithauer BW, Bodosi M. Chordoid glioma of the third ventricle: confirmatory report of a new entity. Hum Pathol 1999; 30: 723-6.
Vanhauwaert DJ, Clement F, Van Dorpe J, Deruytter MJ. Chordoid glioma of the third ventricle. Acta Neurochir (Wien) 2008 Oct 21. [Epub ahead of print].
Wanschitz J, Schmidbauer M, Maier H, Rossler K,Vorkapic P, Budka H. Suprasellar meningioma with expression of glial fibrillary acidic protein: a peculiar variant. Acta Neuropathol (Berlin) 1995; 90: 539-44.
![]()