
Vol. 42, n.º 4, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 4, 2009 |
REVISIONES
José I. López, Ayman Gaafar, Leire Andrés
Servicio de Anatomía Patológica. Hospital
Universitario de Cruces-Osakidetza. Barakaldo, Bizkaia.
joseignacio.lopez@osakidetza.net
RESUMEN
Antecedentes: La médula renal constituye un intrincado sistema de túbulos, vasos sanguíneos e intersticio poco conocido por la mayor parte de los patólogos generales. Métodos: Amplia revisión de la literatura sobre la médula renal y de archivo de toda su patología. Resultados: Se pormenorizan datos de interés para el patólogo sobre el desarrollo normal y patológico, la anatomía microscópica, histología e inmunohistoquímica, la fisiología, la patología de la diferenciación medular (displasia renal multiquística, enfermedades poliquísticas renales autosómicas dominante y recesiva, enfermedad quística medular) la patología inflamatoria (pielonefritis xantogranulomatosa, malacoplaquia), las displasias, y las neoplasias (oncocitoma, tumor oncocítico atípico, carcinoma renal de células cromófobas, carcinoma de los túbulos colectores, carcinoma urotelial, otros carcinomas, fibroma renomedular y tumores metastáticos) de esta topografía. Conclusiones: El conocimiento compendiado de la génesis, del funcionamiento y de la patología medular renal, tanto del desarrollo, como inflamatoria y neoplásica, redundará en un mayor interés por esta zona del riñón que habitualmente pasa desapercibida para el patólogo asistencial.
Palabras clave: Médula renal, embriología, anatomía, fisiología, patología.
SUMMARY
Background: The renal medulla is composed of a complex system of tubules, blood vessels and interstitium, which the general pathologist often are unfamiliar with. Methods: An in depth review of the literature and of material from our archives related to the pathology of the renal medulla was made. Results: Interesting data on normal and abnormal development, microscopic anatomy, histology, immunohistochemistry, physiology, and pathology, including renal medulla differentiation disorders (multicystic renal dysplasia, adult and infantile type polycystic diseases, cystic medullary disease), inflammatory diseases (xanthogranulomatous pyelonephritis, malakoplakia) and neoplasias (oncocytoma, atypical oncocytic tumour, chromophobe renal cell carcinoma, collecting duct carcinoma, urothelial carcinoma, other carcinomas, renomedullary fibroma and metastatic tumours), was reviewed. Conclusions: A comprehensive knowledge of the genesis, function, and pathology of the renal medulla would result in a greater interest being taken in an area often ignored by pathologists.
Keywords: Renal medulla, embryology, anatomy, physiology, pathology.
INTRODUCCIÓN
El interés que despierta el riñón no neoplásico en el patólogo general se focaliza fundamentalmente en el glomérulo. El conjunto de estructuras tubulares que rellena todo el campo del microscopio rodeando glomérulos y vasos sanguíneos constituye un intrincado laberinto mal conocido. La práctica diaria no ayuda a conocer bien la anatomía microscópica de los túbulos renales. Las nefrectomías muestran, por lo general, riñones patológicos, y la presión asistencial no facilita «perder el tiempo» mirando aspectos que no sean relevantes desde el punto de vista del proceso clínico que aqueja al paciente. El riñón de autopsia tampoco es la mejor opción, porque normalmente se trata de personas de edad, con cierta patología vascular o inflamatoria de base que modifica la histología, y porque la autolisis tan propia de este procedimiento hace mella precozmente en las células de los túbulos, haciéndolas prácticamente irreconocibles. Así las cosas, la mejor opción para conocer esta parte del riñón normal es utilizar piezas de nefrectomía postraumática, de accidentes de tráfico por ejemplo, de personas jóvenes. Será necesario también asegurar su perfecta fijación en formol seccionando la pieza precozmente tal y como se aconseja en cualquier texto de patología quirúrgica. Al objeto de tener una visión completa de los túbulos, se debe muestrear la médula renal tanto con cortes longitudinales como transversales.
El interés reciente que han suscitado los túbulos renales viene dado por la concepción actual de que cada tumor renal se origina de una parte concreta de la nefrona (1). Esta idea no es nueva (2), pero ha adquirido especial relevancia a la luz de los últimos hallazgos moleculares que conectan la morfología con diversas alteraciones genéticas más o menos específicas. Al margen del cáncer, otras patologías como las enfermedades quísticas, las hiperplasias/displasias de ductos colectores, y los tumores benignos están pobremente reconocidos entre los patólogos no dedicados a esta área concreta.
En este artículo se revisan de una forma compendiada aspectos normales y patológicos, más o menos conocidos, de la médula renal con la intención de proporcionar un enfoque global diferente a esta topografía tan poco considerada por los patólogos, y a su patología.
EMBRIOLOGÍA
El desarrollo embrionario de las vías excretoras renales está directamente relacionado con el desarrollo de las gónadas y es un capítulo clásico en todos los textos clásicos de embriología (3,4). Dicho de forma esquemática, los túbulos renales son de origen mesodérmico y se desarrollan a partir de los pedúnculos de los somitas, los nefrotomas. En los vertebrados, los túbulos renales están íntimamente asociados con apelotonamientos de vasos sanguíneos finos, los primitivos glomérulos, cuyas paredes endoteliales ya filtran el plasma sanguíneo desde etapas muy tempranas del desarrollo, eliminando los productos nitrogenados de desecho a los túbulos excretores o al celoma adyacente.
El desarrollo de los túbulos excretores progresa en dirección craneo-caudal, diferenciándose antes los túbulos anteriores que los posteriores. Los túbulos que se forman antes (pronefros) son más primitivos y simples en su composición y funcionalismo que los que se forman más tardíamente (mesonefros y metanefros) (4).
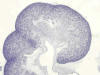
El metanefros es el riñón funcional de la vida postembrionaria en los reptiles, aves y mamíferos (fig. 1). La conversión del magma de células que constituye el metanefros en un sistema de túbulos renales depende del estímulo externo producido en él por parte de la yema ureteral y sus ramificaciones. Esto es así hasta tal punto que la ausencia provocada de manera experimental de botón ureteral en embriones de mamíferos lleva indefectiblemente al no desarrollo del sistema de túbulos renales.
ANATOMÍA MICROSCÓPICA, HISTOLOGÍA E INMUNOHISTOQUÍMICA
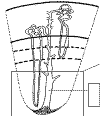
La médula renal está formada por estructuras cónicas, las pirámides renales, cuyas bases se sitúan en la unión córtico-medular (fig. 2). Cada pirámide renal, junto con la parte cortical que la cubre, constituye el lóbulo renal. La base del lóbulo renal es la cápsula renal y la punta del mismo es la papila con la lámina cribosa asomando a la pelvis renal. El número de papilas que contiene cada riñón es variable, oscilando entre 5 y 11, siendo 8 la cantidad más constante. Un estudio clásico sobre 375 riñones (5) indica que el número de conductos colectores de Bellini que conforman la lámina cribosa de cada papila oscila entre 116 y 776. Los límites laterales de cada lóbulo renal son las columnas de Bertin, estructuras residuales que representan la fusión de los lóbulos primitivos dentro del blastema metanéfrico primitivo. A ese nivel se sitúan las arterias interlobulares. Cada lóbulo renal contiene un solo ducto colector, a donde drenan multitud de glomérulos, tanto corticales como yuxtamedulares.
El ducto colector de Bellini tiene una parte cortical y otra medular y recorre prácticamente todo el espesor del tejido renal, desde la cápsula hasta la papila (fig. 3). El túbulo urinífero, desde su salida de la cápsula de Bowman hasta su desembocadura en el ducto colector de Bellini, tiene una longitud de entre 5 y 7 mm (6). Las nefronas corticales tienen la mayor parte de su recorrido, incluyendo un asa de Henle corta, en la corteza. Sólo una parte pequeña de la misma se introduce en la parte más externa (yuxtacortical) de la médula. La nefronas yuxtamedulares, por el contrario, tienen un asa de Henle larga, que está toda ella incluida en la médula. Por lo tanto, mientras la médula renal sólo contiene asas de Henle y ductos colectores de Bellini, la corteza incluye, además, la totalidad de los túbulos contorneados proximales y distales.
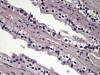
El asa de Henle tiene una parte gruesa y otra fina (figs. 4 y 5). La parte gruesa contiene células de citoplasma eosinófilo y cuboideo aplanado. La parte fina es muy delicada, con epitelio aplanado simple, y puede confundirse con capilares en el corte transversal. La pared es casi inaparente y el núcleo, muy notorio en comparación con la pared, hace prominencia en la luz a la manera que lo hacen las células endoteliales en los capilares. Ambas porciones del asa de Henle muestran intensa positividad con panqueratinas del tipo AE1/AE3 y con citoqueratina 7 (figs. 6 y 7). Las citoqueratinas 8, 18, el EMA y la e-cadherina presentan inmunotinción leve.

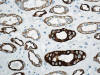
Los ductos colectores están tapizados por dos tipos diferentes de células: las células principales y las células intercalares (figs. 5 y 8). Ambas células son prácticamente indistinguibles en cortes de hematoxilina-eosina. Las células principales tienen un cilio apical inmóvil y las intercalares abundantes microvilli apicales y mitocondrias. El cilio apical de las células principales es un sensor local del flujo de líquidos y de sus contenidos. Este cilio contiene dos proteínas asociadas a membrana denominadas policistina-1 y policistina-2. Las células principales y las intercalares muestran positividad intensa con citoqueratinas 7, 8, 18, panqueratina de tipo AE1/AE3, EMA, y e-cadherina (figs. 9 y 10). Las células principales son positivas con c-Kit (fig. 11). Es importante recordar que el túbulo colector tiene una parte de su recorrido en la corteza renal, lugar en donde son especialmente abundantes las células intercalares (7). Esta es la razón por la cual los tumores de la nefrona distal también pueden localizarse periféricamente en el riñón, abombando la superficie convexa del mismo, en plena corteza renal, mimetizando a aquellos otros tumores provenientes de la nefrona proximal. Las células intercalares son células «oscuras» en las imágenes del microscopio electrónico debido a su alto contenido en organelas intracitoplásmicas, especialmente mitocondrias. Tienen un alto contenido en anhidrasa carbónica, motivo por el cual han sido relacionadas con la acidificación de la orina.
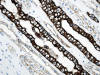
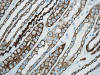

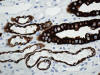
La otra estructura incluida en la médula renal son los vasos rectos (vasa recta). Éstos se originan de la arteriola eferente del glomérulo, descienden en la médula como capilares arteriales de endotelio continuo, dan la vuelta y ascienden como capilares venosos de endotelio discontinuo hasta alcanzar la unión córtico-medular, y allí desembocan finalmente en las venas interlobulares. En todo su trayecto, descendente y ascendente, las vasos rectos se sitúan en íntima relación con las asas de Henle y con los ductos colectores, presentando inmunotinción positiva con factor VIII y con CD31 (fig. 12).
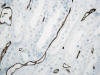
Por último, entre los vasos rectos y las asas de Henle se localizan las células intersticiales, que se disponen de forma ordenada y paralela semejando los peldaños de una escalera de mano (6).
FISIOLOGÍA
La fisiología de la médula renal está ligada íntimamente a la absorción o excreción de líquidos y solutos, y participa de manera crucial en la regulación de la presión arterial. El asa de Henle reabsorbe el 15% del H2O filtrada y el 25% del NaCl, K+, Mg+ Ca++, HCO3– (8). La reabsorción del H2O depende del gradiente osmótico entre la luz y el intersticio, y ocurre exclusivamente en la parte descendente del asa ya que la parte ascendente de la misma es impermeable a este elemento. La reabsorción de los solutos citados se realiza gracias a una bomba ATPasa dependiente. Los diuréticos tiazídicos inhiben este mecanismo invirtiendo la osmolaridad y favoreciendo con ello la eliminación del agua (9) y la regulación de la tensión arterial.
Los ductos colectores de la zona medular interna es la zona donde termina de definirse la concentración de la orina y es donde se realiza la reabsorción de agua mediada por vasopresina (9). Las células principales de los ductos colectores reabsorben agua y Na+ y excretan K+ mediante una bomba de Na-K ATPasa dependiente. Las células intercalares excretan H+ o CO3H– y reabsorben K+.
Las variaciones en los niveles de Na+ en la parte distal de la nefrona determinan la mayor o menor producción de renina en el aparato yuxtaglomerular. La renina cataboliza el paso de angiotensina a angiotensina I. Ésta última se convierte localmente en angiotensina II. Esta cascada de acontecimientos tiene un triple efecto: contracción de las células musculares lisas, aumento en la secreción de aldosterona en las glándulas adrenales, y reabsorción aumentada de Na+ para elevar la presión arterial (10).
Además de las funciones tradicionalmente conocidas, las angiotensinas producidas localmente participan de manera autocrina o paracrina en múltiples procesos relacionados con el crecimiento, la diferenciación, y la proliferación celulares, así como con la angiogénesis (11-13). La enzima convertidora de angiotensina (ECA) es un metalopéptido que convierte la angiotensina I en la angiotensina II, un potente vasopresor. Clásicamente se ha considerado a esta enzima como el regulador clave sobre el que actuar farmacológicamente para modular la hipertenión arterial. Sin embargo, el descubrimiento reciente de la enzima convertidora de angiotensina 2 (ECA2), una enzima que convierte en varios tejidos la angiotenina II en el vasodilatador ANG 1-7, ha puesto en evidencia la tremenda complejidad de este sistema (14,15). ECA y ECA2 se expresan en los túbulos renales y juegan un papel muy importante en la regulación del sistema renina-angiotensina local (16) y su disfunción ha sido estudiada por varios autores en diversas enfermedades renales no neoplásicas (13,17-19) y, por el que suscribe este artículo, junto con otros, en diversos carcinomas de células renales y en oncocitomas (20-22).
Las células intersticiales de la médula renal son de naturaleza fibroblástica y contribuyen a mantener la estructura de la zona. Estas células producen la eritropoyetina, y se ha sugerido también que segregan prostaglandinas.
ANATOMIA PATOLÓGICA
Anomalías en la diferenciación medular renal
Constituyen un conjunto heterogéneo de enfermedades esporádicas o hereditarias que cursan con diversos grados y formas de transformación quística tubular. Varias de ellas afectan a la corteza, o a la corteza y a la médula. En esta revisión se incluyen sólo aquellas patologías que afectan selectivamente a los ductos colectores.
Displasia Renal Multiquística
Es una patología de origen oscuro, unilateral, más frecuente en varones y en el lado izquierdo, que cursa con hipertensión arterial. La mayor parte de los casos son esporádicos, aunque existen raros casos hereditarios.
Es una patología que está causada por alteraciones mal conocidas en la diferenciación del metanefros que llevan a una mala diferenciación córtico-medular y a un mal desarrollo de los cálices renales (23). Se observan multitud de quistes originados en los ductos colectores primitivos que muestran morfología y tamaño variables y que distorsionan completamente la morfología renal (fig. 13). Se asocia a oclusión pielo-calicial o a atresia ureteral.
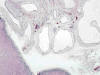
Desde el punto de vista morfológico los ductos colectores tienen una apariencia displásica, existen escaso desarrollo de las nefronas, con escasa o nula presencia de asas de Henle y de vasos rectos, debido a la falta de inducción de la yema ureteral. Los túbulos y quistes distribuidos de forma irregular están inmersos en un estroma fibroso, con presencia de nódulos de blastema metanéfrico indiferenciado y nidos de cartílago maduro.
Enfermedad Poliquística Renal Autosómica Dominante
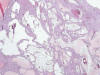
Esta enfermedad es el resultado de la mutación de 2 genes, PKD1, localizado en el cromosoma 16, y PKD2, localizado en el cromosoma 4, que codifican respectivamente la poliquistina 1 y la 2, proteínas relacionadas con el cilio inmóvil de las células principales del ducto colector (6). La alteración de PKD1 es la responsable de hasta el 90% de los casos de la enfermedad. La enfermedad se denomina también enfermedad poliquística del adulto y se hace patente hacia la cuarta década de la vida. Típicamente cursa con hipertensión arterial e infecciones urinarias frecuentes. Existe una forma clásica de la enfermedad y otra de comienzo precoz, en la infancia o en la adolescencia.
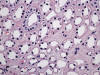
La transformación quística progresiva de los ductos colectores lleva a desarrollar riñones enormes con infinidad de quistes de tamaño variable, con contenido líquido (fig. 14). El parénquima renal localizado entre los quistes desarrolla fenómenos de atrofia por compresión. Se observa también dilatación quística del espacio de Bowman.
Enfermedad Poliquística Renal Autosómica Recesiva
La enfermedad aparece como consecuencia de la mutación del gen PKHD1, localizado en el cromosoma 6, que codifica una proteína denominada fibroquistina. La enfermedad se conoce también como enfermedad poliquística infantil, aunque eventualmente algunos casos pueden hacerse sintomáticos en la adolescencia o incluso en la vida adulta. Existe una forma clásica de la enfermedad y otra asociada a fibrosis hepática congénita (23). La transformación quística de los ductos colectores afecta también al trayecto cortical de los mismos, de tal forma que todo el espesor del riñón está afectado por quistes de tamaño pequeño. Los quistes en la zona cortical del ducto colector tienden a ser alongados o fusiformes, mientras que en la zona medular son redondeados (fig. 15).
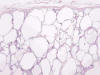
Enfermedad Quística Medular
Se han descrito dos formas clínicas de esta enfermedad, que afectan selectivamente a los ductos en la médula renal, el riñón en esponja y el complejo enfermedad quística medular-nefronoptisis familiar (23).
El riñón en esponja es relativamente frecuente y afecta a los túbulos colectores en la zona más distal de los mismos, deformando las papilas renales. Con frecuencia afecta sólo a una o dos pirámides renales, o a un solo riñón. Es una enfermedad de aparición esporádica, aunque se han descrito casos de asociación familiar en gemelos o en relación con otras enfermedades como la hemihipertrofia congénita, Marfan, o Elhers-Danlos. Clínicamente cursa con litiasis renal frecuente y hematuria.
La enfermedad quística medular y la nefronoptisis familiar se describieron en un principio de forma separada y posteriormente se han reunido bajo el mismo síndrome debido a sus hallazgos comunes desde el punto de vista clínico y patológico. La enfermedad quística medular es autosómica dominante y comienza en la vida adulta y la nefronoptisis familiar es de inicio más precoz, en la adolescencia, y se hereda de forma autosómica recesiva.
En ambas se observa atrofia tubular distal con engrosamiento o duplicación de la membrana basal en los túmulos que sobreviven. Existe dilatación quística de los túbulos a nivel de la unión córtico-medular.
Nefritis Intersticiales
La mayor parte de las nefritis túbulo-intersticiales afectan a la corteza renal y exceden al objetivo de este trabajo. El papel de los fibroblastos intersticiales y de las células dendríticas en esta patología concreta ha sido objeto de revisiones recientes (24) y abre innumerables vías de investigación. No obstante, se enumeran brevemente los hallazgos intersticiales medulares que pueden verse en cada uno de los grupos sindrómicos. Se hace más hincapié en la pielonefritis xantogranulomatosa y la malacoplaquia porque afectan preferentemente a la médula renal y porque con frecuencia se pueden confundir con procesos neoplásicos.
La nefritis intersticial aguda tiene una prevalencia en torno al 1% de de todas las biopsias renales realizadas para el estudio de hematuria o proteinuria. Se asocia sobre todo a la toma de medicamentos (anti-inflamatorios no esteroideos, antibióticos, sulfonamidas, alopurinol…) aunque se han descrito casos secundarios a sarcoidosis e infecciones. Histológicamente se observa edema e infiltrado inflamatorio intersticial por linfocitos y células plasmáticas. También pueden verse granulomas.
La nefritis intersticial crónica describe un grupo de enfermedades que afectan a los túbulos renales y al intersticio. Es más frecuente que la forma aguda. Histológicamente se caracteriza por atrofia y dilatación tubular, fibrosis intersticial, e infiltrados inflamatorios crónicos con eosinófilos acompañantes. Los glomérulos están preservados en un principio, pero pueden afectarse secundariamente a lo largo de la enfermedad. Entre las causas que la producen están: ciertas enfermedades hereditarias (enfermedad poliquística, enfermedad quística medular), alteraciones metabólicas (hipercalcemia, nefrocalcinosis, hiperoxaluria, hipopotasemia, hiperuricemia, cistinosis…), fármacos y tóxicos (analgésicos, cadmio, plomo, litio, ciclosporina, cisplatino…), alteraciones inmunológicas (rechazo renal, lupus eritematoso sistémico, granulomatosis de Wegener, vasculitis, síndrome de Sjögren, sarcoidosis…), alteraciones hematológicas (mieloma múltiple, linfomas, hemoglobinuria paroxística nocturna…), infecciones, etc.
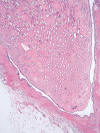
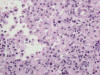
La pielonefritis crónica xantogranulomatosa es una variedad especial de inflamación crónica del parénquima renal que simula radiológica y macroscópicamente un tumor renal (25). Se produce por la respuesta anormal del huésped frente a una infección bacteriana, comúnmente E. Coli o Proteus Mirabilis, que lleva a la destrucción del parénquima renal y a su sustitución por macrófagos xantomizados. La inflamación borra la unión córtico-medular formando masas irregulares, puede incluso sobrepasar la cápsula renal y afectar a la grasa perinéfrica, lo cual aumenta la confusión con un proceso maligno. Microscópicamente se observan histiocitos espumosos (fig. 16) mezclados con fibrosis reactiva e inflamación linfoplasmocítica y polimorfonuclear variable. La inflamación xantogranulomatosa no es exclusiva del tracto urinario, pudiendo encontrarse en otros órganos. En ocasiones, pueden coexistir inflamación xantogranulomatosa y tumor, lo cual suele complicar suele complicar el reconocimiento de este último, normalmente ahogado en un contexto de inflamación abigarrado (26).
La malacoplaquia renal es una reacción inflamatoria crónica infrecuente asociada a la infección por E. Coli que afecta con preferencia al tracto urinario, sobre todo a la vejiga. Su patogenia es superponible a la de la pielonefritis xantogranulomatosa. Se ha visto un aumento de incidencia en pacientes inmunocomprometidos (27). La malacoplaquia también puede simular un tumor renal. El cuadro histológico característico incluye la presencia de inclusiones basofílicas (cuerpos de Michaelis-Gutmann), que corresponden a componentes bacterianos fagocitados, en el citoplasma de macrófagos de citoplasma eosinofílico; los denominados histiocitos de von Hansemann.
Al igual que en el caso anteriormente comentado, la malacoplaquia no es una entidad exclusiva del tracto urinario, pudiendo encontrarse en otros órganos. También pueden coexistir tumor e inflamación xantogranulomatosa.
Hiperplasias y Displasias
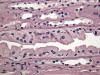
En determinadas circunstancias, por ejemplo algunas litiasis, se puede observar una hiperplasia del epitelio de la parte más distal de los túbulos colectores (fig. 17). En las formas más extremas las papilas renales contienen túbulos colectores dilatados y rellenos de células, sin casi luz aparente, y los túbulos se encuentran apretados unos a otros, sin estroma interviniente aparente. Suele acompañarse de hiperplasia del urotelio adyacente reflejando una respuesta conjunta frente a la misma noxa.
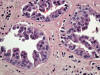
La células del túbulo colector pueden mostrar cambios displásicos (fig. 18), con dismorfias, pseudoestratificación e hipercromatismo nuclear casi siempre en relación con carcinomas uroteliales de la pelvis renal adyacente.
Neoplasias
Oncocitoma Renal
El oncocitoma renal es un tumor benigno que constituye alrededor del 5% de las neoplasias renales del adulto y que se origina a partir de las células intercalares (1). Con frecuencia es un tumor asintomático que se descubre casualmente al estudiar el retroperitoneo por otras causas. Se trata de un tumor formado por «oncocitos», células de morfología poliédrica con citoplasmas densamente eosinófilos granulares y núcleos cromáticos centrales (fig. 19). El oncocitoma es un tumor bien delimitado, no encapsulado, de color marrón oscuro, que muestra a veces una cicatriz fibrosa central típica. Las células del oncocitoma contienen abundantes mitocondrias y son negativas con la técnica del hierro coloidal.
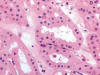
El diagnóstico diferencial debe incluir sobre todo al carcinoma renal de células cromófobas y al carcinoma renal de células claras, variante de células granulares (28), debido al diferente pronóstico que comportan estas otras entidades. Aparte de las características puramente histológicas, la inmunohistoquímica y los estudios moleculares pueden ayudar decisivamente a distinguirlos. No obstante, algunos casos aislados plantean grandes dudas. Desde el punto de vista inmunohistoquímico, el oncocitoma renal es positivo con CD15, e-cadherina, parvalbúmina, c-Kit, y con la citoqueratina 20. La citoqueratina 7 puede ser positiva de manera focal (29). Estudios recientes muestran positividad para claudina 8 (30) y para ciclina D1 (31). El oncocitoma muestra pérdidas de material en los cromosomas 1 y 14, así como alteraciones del ADN mitocondrial.
Tumor Oncocítico Atípico
El término tumor oncocítico atípico se acuñó para resolver el conflicto provocado por la constatación clínica de inesperadas metástasis a distancia en oncocitomas renales, por un lado, y por la curación tras muchos años de seguimiento de algunos carcinomas renales de células cromófobas, por otro. En ambos casos, para bien o para mal, se trata de comportamientos clínicos anómalos en los que el patólogo pudo ayudar poco. Histológicamente son tumores que comparten características mixtas, a medio camino entre el oncocitoma y el carcinoma renal de células cromófobas. El autor ha realizado una revisión reciente de esta problemática (32).
Carcinoma Renal de Células Cromófobas
El carcinoma renal de células cromófobas, descrito por primera vez por Thoenes y cols. (33) en 1985, y tiene su origen también en las células intercalares de los túbulos colectores (34). Existe una variedad de este tumor denominada eosinofílica, también descrita por este mismo autor alemán unos años más tarde (35). El carcinoma renal de células cromófobas supone aproximadamente el 5% de todos los tumores renales de adultos y, en términos generales, muestra mejor pronóstico que la variedad de células claras, aunque éste está también sujeto al estadio y al grado en el momento del diagnóstico (36). No obstante, al igual que éste, también puede presentar transformación sarcomatoide (37,38).
Histológicamente es un tumor bien conocido constituido por células poliédricas, de aspecto «vegetal», con citoplasmas eosinófilos débilmente granujientos y halo claro perinuclear (fig. 20). Una revisión reciente a cargo de Amin y cols (38) destaca de estos tumores que el cambio sarcomatoide, la presencia de necrosis y el pT alto al diagnóstico son factores histológicos predictivos de comportamiento agresivo. Quizá la variedad eosinofílica pueda plantear ciertos problemas de diagnóstico diferencial en cortes de hematoxilina-eosina con el oncocitoma renal. Así, la forma convencional de este tumor está compuesta por células poliédricas de citoplasma amplio levemente granujiento, eosinófilo, con membrana citoplásmica prominente, y los núcleos centrales, con halo perinuclear (1). El carcinoma de células cromófobas es positivo con la técnica de hierro coloidal y muestra abundantes microvesículas en el citoplasma cuando se estudia con el microscopio electrónico. Desde el punto de vista genético, este tumor se caracteriza por pérdidas múltiples de cromosomas (Y, 1, 2, 6, 10, 13, 17, 21), lo cual es un hallazgo típico según algunos autores (34).
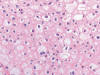
Desde el punto de vista inmunohistoquímico, el carcinoma renal de células cromófobas remeda el patrón de inmunotinción de la célula de la que supuestamente se origina, mostrando positividad con citoqueratina 7, EMA, e-cadherina, c-Kit, y parvalbúmina (29). Estudios recientes muestran positividad para Claudina 7 (30).
Carcinoma de los Ductos Colectores
El carcinoma de los ductos colectores, o carcinoma de los ductos de Bellini, es una forma poco frecuente de carcinoma renal, aconteciendo en aproximadamente el 1% de los casos de carcinoma renal en adultos. Característicamente tiene mal pronóstico, como demuestran Tokuda y cols. (39) en la mayor serie recogida hasta la fecha en la literatura con 81 pacientes.
Macroscópicamente es un tumor que forma masas blanquecinas, no amarillentas, de consistencia firme, que tienden a afectar fundamentalmente la médula renal, y en este sentido puede distinguirse del resto de carcinomas renales. Histológicamente, el carcinoma de los ductos colectores está constituido por múltiples estructuras ductales de contorno irregular inmersas en un estroma fibroso, a veces incluso desmoplásico (40) (fig. 21). Las células neoplásicas muestran atipia intensa, pleomorfismo, hipercromatismo y mitosis, con elementos frecuentes «en tachuela» o «en anillo de sello» (41). Desde el punto de vista histológico debe distinguirse del carcinoma renal papilar y del carcinoma urotelial de alto grado (39,40).
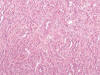
Desde el punto de vista inmunohistoquímico, el carcinoma de los ductos colectores es típicamente positivo con EMA y citoqueratinas de alto peso molecular. En ocasiones resulta positivo con citoqueratina 7. CD10, CD117 y citoqueratina 20 son negativos (29). Las alteraciones genéticas encontradas son variadas y poco específicas.
Existen en la literatura varios ejemplos de carcinomas renales con características intermedias entre carcinoma de los ductos colectores y carcinoma urotelial (42,43), reflejando la proximidad histogenética existente entre ambos tumores.
Carcinoma Urotelial
El carcinoma urotelial a este nivel comparte las mismas características epidemiológicas que el carcinoma urotelial del ureter o de la vejiga. La mayor parte de estos tumores crecen de forma exofítica en la luz de la pelvis renal, pero otros, generalmente de alto grado, invaden directamente la médula renal induciendo abundante desmoplasia en el estroma. En ocasiones se puede observar la extensión ascendente del tumor a través de los túbulos colectores. En estos casos, en vecindad, puede observarse cierto grado de hiperplasia y/o de displasia en el epitelio del túbulo colector.
Histológicamente, el carcinoma urotelial de la pelvis renal es superponible al de la vejiga urinaria. En los casos de tumores de alto grado, indiferenciados, que invaden la médula renal, el diagnóstico diferencial con el carcinoma de los ductos colectores puede ser dificultoso.
Otros carcinomas
Hace aproximadamente una década la literatura especializada comienza a recoger descripciones de casos sueltos, o de pequeñas series, de ciertos tumores renales presumiblemente relacionados con la nefrona distal (44-48). Se trataba de tumores de morfología peculiar, tubular, cuboidea, a veces fusocelular, y se acuñó para referirse ellos el término de «loopoma» (49). Rakozy y cols. (50) hacen mención a estos tumores con el nombre de carcinoma tubular, mucinoso y fusocelular (fig. 22), nombre que aún permanece. Sin embargo, se ha demostrado hoy en día que estos tumores son una variedad histológica peculiar de carcinoma renal papilar, con el cual comparte las alteraciones genéticas típicas (51).
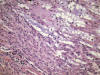
Algo similar ha ocurrido con el carcinoma renal túbulo-quístico (fig. 23), entidad considerada en la descripción original a cargo de Mac Lennan y cols. en 1997 (52) como la variedad de bajo grado del carcinoma de los ductos colectores y que ha pasado a engrosar la lista de recién llegados al grupo del carcinoma renal papilar (53).
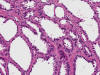
En la literatura se pueden encontrar casos aislados de tumores mal conocidos, derivados de la nefrona distal, que recapitulan casi todas las variedades histológicas (oncocitoma, carcinoma renal de células cromófobas y carcinoma de los ductos colectores) (48).
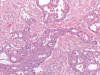
El carcinoma renal medular es una variedad histológica de carcinoma renal que se observa en pacientes de raza negra con rasgo drepanocítico (HbSA y HbSC) (54). Curiosamente, la anemia drepanocítica (HbSS) no se asocia al tumor. Es un tumor agresivo, con muy cortas supervivencias, que se desarrolla en pacientes jóvenes a partir de los túbulos colectores o del epitelio de las papilas renales (55). Es un buen ejemplo de tumor infiltrante, constituido por células atípicas dispuestas en patrones de crecimiento variados (fig. 24), simulando en ocasiones el carcinoma adenoide quístico, o incluso el tumor del saco vitelino.
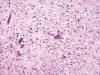
Fibroma Renomedular
Es un tumor poco frecuente, asintomático, que se descubre en ocasiones como hallazgo autópsico casual. Deriva de las células intersticiales de la médula renal. Son tumores pequeños, de menos de 1 cm que se localizan en las porciones medias de las pirámides renales. Microscópicamente están constituidos por células fusiformes o estrelladas inmersas en un estroma edematoso que engloba también túbulos medulares (fig. 25). Pueden presentar hialinización y, menos frecuentemente amiloide (56).
Tumores Metastásicos
Las metástasis de otros tumores en la médula renal son raras. Entre ellas, los orígenes más frecuentes son el pulmón, la mama, y el tracto digestivo (57). Pueden adoptar un patrón macroscópico infiltrante o expansivo, y normalmente son múltiples y bilaterales.
CONCLUSIONES
La médula renal es una topografía poco reconocida por los patólogos que, sin embargo, es el origen de muy diversas patologías, tanto del desarrollo, como inflamatorias o neoplásicas. El conocimiento más detallado de la embriología de la médula renal, de su anatomía microscópica y de su fisiología ayudará a entender mejor la patología y a integrar estos datos con la clínica. Esta revisión pretende compendiar en pocas páginas los puntos más importantes desde una aproximación multidisciplinar.
BIBLIOGRAFÍA
López-Beltrán A, Scarpelli M, Montironi R, Kirkali Z. 2004 WHO classification of the renal tumors of the adults. Eur Urol 2006; 49: 798-805.
Holthöfer H, Miettinen A, Paasivuo R, Lehto VP, Linder E, Alfthan O, Virtanen I. Cellular origin and differentiation of renal carcinomas. A fluorescence microscopic study with kidney-specific antibodies, antiintermediate filament antibodies , and lectins. Lab Invest 1983; 49: 317-26.
Willis RA. The Borderland of Embryology and Pathology. Butterworth and Co. Ltd., London 1958.
Balinski BI. Introducción a la Embriología, 5.ª Ed., Ediciones Omega S.A. Barcelona, 1983.
Williams PL, Warwick R. Anatomia de Gray, 36ª Edición, Tomo 2, Churchill-Livingstone, Edimburgo, 1996.
Kierszenbaum AL. Histology and Cell Biology. An Introduction to Pathology, 2nd ed., Mosby Elsevier, Philadelphia, 2007.
Clapp WL, Croker BP. Kidney. In Mills SE, ed, Histology for Pathologists, Chapter 34, 2nd edition, Lippincott Williams-Wilkins, Philadelphia, 2007 pp 839-907.
Guyton AC, Hall JE. Tratado de Fisiología Médica, 9.ª Edición, Interamericana-McGraw-Hill, Nueva York, 1997.
Janech MG, Raymond JR, Arthur JM. Proteomics in renal research. Am J Physiol Renal Physiol 2007; 292: F501-12.
Kumar V, Abbas AK, Fausto N, Mitchell RN. Robbins Patología Humana, 8ª Edición, Elsevier Epaña, 2008.
Deshayes F, Nahmias C. Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab 2005; 16: 293-9.
Paul M, Mehr AP, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 2006; 86:747-803.
Kobori H, Masaomi N, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59:251-87.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robinson K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000; 87: 1-9.
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000; 275: 33238-43.
Danylczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res 2006; 98: 463-71.
Metzger R, Bohle RM, Pauls K, Eichner G, Alhenc-Gelas F, Danilov SM, Franke FE. Angiotensin-converting enzyme in non-neoplastic kidney diseases. Kidney Int 1999; 56: 1442-54.
Lely AT, Hamming I, van Goor H, Navis GJ. Renal ACE2 expression in human kidney disease. J Pathol 2004; 204: 586-93.
Mizuiri S, Hemmi H, Arita M, Ohashi Y, Tanaka Y, Miyagi M, Sakai K, Ishikawa Y, Shibuya K, Hase H, Aikawa A. Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am J Kidney Dis 2008; 51: 613-23.
Varona A, Blanco L, López JI, Gil J, Agirregoitia E, Irazusta J, Larrinaga G. Altered levels of acid, basic, and neutral peptidase activity and expression in human clear cell renal cell carcinoma. Am J Physiol-Renal Physiol 2007; 292: 780-8.
Larrinaga G, López JI, Casis L, Blanco L, Gil J, Agirregoitia E, Varona A. Cystinil aminopeptidase activity is decreased in renal cell carcinoma. Regul Pept 2007; 144: 56-61.
Blanco L, Larrinaga G, Pérez I, López JI, Gil J, Agirregoitia E, Varona A. Acid, basic and neutral peptidases present different profiles in chromophobe renal cell carcinoma and in oncocytoma. Am J Physiol-Renal Physiol 2008; 294: 850-8.
Lacson A, Bernstein J, Ridson RA, Gilbert-Barness E. Renal System. Kidneys and Urinary Tract, Chapter 27, part 1. En Potter’s Pathology of the Fetus, Infant and Child, Gilbert-Barness E Ed., vol 2, 2nd Edition, Mosby-Elsevier, Philadelphia, 2007, pp 1281-1344.
Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol 2008; 247-62.
Malek RS, Elder JS. Xanthogranulomatous pyelonephritis: a clinical analysis of 26 cases and of the literature. J Urol 1978; 119: 589-93.
López JI, Elizalde JM, Calvo MA. Xanthogranulomatous cholecystitis associated with gallbladder adenocarcinoma. A clinicopathologic study of 5 cases. Tumori 1991; 77: 358-360.
Biggar WD, Keating A, Bear RA. Malakoplakia: evidence for an acquired disease secondary to immunosuppression. Transplantation 1981; 31: 109-12.
Abrahams NA, Tamboli P. Oncocytic renal neoplasms: Diagnostic considerations. Clin Lab Med 2005; 25: 317-39.
Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical markers in the differential diagnosis of renal neoplasms. Clin Lab Med 2005; 25: 247-57.
Lechpammer M, Sabo E, Yakirevich E, Sciandra K, Greaves W, Tavares R, Noble L, Resnick MB, Wang LJ. Claudins 7 and 8 expression separate chromophobe renal cell carcinoma and oncocytoma. Mod Pathol 2008; 21: 186A.
Sukov WR, Carlson AW, Lager DJ, Ketterling RP, Cheville JC. Cyclin D1 overexpression and CCND1 rearrangement in renal oncocytoma: Frequency, clinicopathologic features and utility in differentiation from chromophobe renal cell carcinoma. Mod Pathol 2008; 21: 182A.
Lopez JI, Ugalde A, Ortega FJ, Vilanova JR. Neoplasias oncocíticas renales. Revisión crítica de un problema diagnóstico no resuelto. Rev Esp Patol 2008; 41: 249-59.
Thoenes W, Störkel S, Rumpelt HJ. Human chromophobe cell renal carcinoma. Virchows Arch (B) 1985; 48: 207-17.
Cheng L, Zhang S, MacLennan GT, Lopez-Beltrán A, Montironi R. Molecular, and cytogenetic insights into the pathogenesis, classification, differential diagnosis, and prognosis of renal epithelial neoplasms. Hum Pathol 2009; 40: 10-29.
Thoenes W, Störkel S, Rumpelt HJ, y cols. Chromophobe renal cell carcinoma and its variants. A report on 32 cases. J Pathol 1988; 155: 277-87.
Nese N, Paner GP, Mallin K, Ritchey J, Stewart A, Amin MB. Renal cell carcinoma: Assessment of key pathologic prognostic parameters and patient characteristics in 47909 cases using the National Cancer Data Base. Ann Diagn Pathol 2009; 13: 1-8.
Nagashima Y, Okudela K, Osawa A y cols. Chromophobe renal cell carcinoma with sarcomatoid change. A case report. Pathol Res Pract 2000; 196: 647-51.
Amin MB, Paner GP, Alvarado-Cabrero I, Young AN, Stricker HJ, Lyles RH, Moch H. Chromophobe renal cell carcinoma: Histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 145 cases. Am J Surg Pathol 2008; 32: 1822-34.
Tokuda N, Naito S, Matsuzaki O, Nagashima Y, Ozono S, Igarashi T. Collecting duct (Bellini duct) renal cell carcinoma: A Nationwide survey in Japan. J Urol 2006; 176: 40-3.
Kobayashi N, Matsuzaki O, Shirai S, Aoki I, Yao M, Nagashima Y. Collecting duct carcinoma of the kidney: an immunohistochemical evaluation of the use of antibodies for differential diagnosis. Hum Pathol 2008; 39: 1350-9.
Li M, Vuolo MA, Weidenheim KM, Minsky LS. Collecting-duct carcinoma of the kidney with prominent signet ring cell features. Mod Pathol 2001; 14: 623-8.
Orsola A, Trías I, Raventós CX, Español I, Cecchini L, Orsola I. Renal collecting (Bellini) duct carcinoma displays similar characteristics to upper tract urothelial cell carcinoma. Urology 2005; 65: 49-54
Meastroni U, Ferretti S, Dinale F, Froio E, Pilato FP, Ciuffreda M, Mellissari M, Cortellini P. A renal cancer with intermediate characteristics between collecting (Bellini) duct carcinoma and urothelial carcinoma: case report review of the literature. Tumori 2006; 92: 545-8.
Parwani AV, Husain AN, Epstein JI, Bruce Beckwith J, Argani P. Low-grade myxoid renal epithelial neoplasms with distal nephron differentiation. Hum Pathol 2001; 32: 506-12.
Otani M, Shimizu T, Serizawa H, Ebihara Y, Nagashima Y. Low-grade renal cell carcinoma arising from the lower nephron: A case report with immunohistochemical, histochemical, and ultrastructural studies. Pathol Int 2001; 51: 954-60.
Leroy X, Aubert S, Gosselin B. Low-grade myxoid renal epithelial neoplasms with distal nephron differentiation: A distinct clinicopathtologic entity? Hum Pathol 2002; 33: 574-5.
Hara N, Kawaguchi M, Koike H, Tsutsui T, Takagi R, Takahashi K. Low-grade renal epithelial tumor originating from the distal nephron. Int J Urol 2004; 11: 68-73.
Lindgren V, Paner GP, Flanigan RC, Clark JI, Campbell SC, Picken MM. Rernal tumor with overlapping distal nephron morphology and karyotype. Arch Pathol Lab Med 2004; 128: 1274-8.
Hes O, Hora M, Perez-Montiel DM, y cols. Spindle and cuboidal renal cell carcinoma (loopoma): 10 case reports. Cas Lek Cesk 2004; 143: 169-73.
Rakozy C, Schmahl GE, Bogner S, Storkel S. Low-grade tubular-mucinous renal neoplasms: morphologic, immunohistochemical, and genetic features. Mod Pathol 2002; 15: 1162-71.
Ugalde A, López JI. El espectro del carcinoma renal papilar. Actas Urol Esp 2008; 32: 799-805.
MacLennan GT, Farrow GM, Bostwick DG. Low-grade collecting duct carcinoma of the kidney: Report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology 1997; 50: 679-84.
Yang Xj, Zhou M, Hes O, Shen S, Li R, López JI, Shah RB, Yang Y, Chuang ST, Lin F, Tretiakova MM, Kort EJ, The BT. Tubulocystic carcinoma of the kidney. Clinicopathologic and molecular characterization. Am J Surg Pathol 2008; 32: 177-87.
Herring JC, Schmetz MA, Digan AB, Young ST, Kaloo NB. Renal medullary carcinoma: a recently described highly aggressive renal tumor in young black patients. J Urol 1997; 157: 2246-7.
Davis CJ, Mostofi FK, Sesterhenn IA. Renal medullary carcinoma: the seventh sickle nephropathy. Am J Surg Pathol 1995; 19: 1-11.
Murphy WM, Grignon DJ, Perlman EJ. Tumors of the Kidney, Bladder, and Related Urinary Structures. Armed Forces Institute of Pathology, 4th Series, Fascicle 1, Washington DC, 2004.
Bracken RB, Chica G, Johnson DE, Luna M. Secondary renal neoplasms: an autopsy study. South Med J 1979; 72: 806-7.
![]()