
Vol. 35, n.º 1, 2002
| REVISTA
ESPAÑOLA DE Vol. 35, n.º 1, 2002 |
Carmen Morales Bastos
Médico adjunto del Departamento de Anatomía Patológica del Hospital Universitario La Paz y Profesora Asociada de Anatomía Patológica en la Facultad de Medicina de la Universidad Autónoma de Madrid.
Trabajo basado en la Conferencia «Neuropatología Perinatal» de C. Morales Bastos en el I Congreso Virtual Hispanoamericano de Anatomía Patológica (www.conganat.org/icongreso), 15-5 al 7-7 de 1997.
INTRODUCCIÓN
Los dos últimos trimestres de la gestación y los primeros meses de vida son un periodo crítico en el que se produce el 80% del crecimiento del Sistema Nervioso Central (SNC) (21). El desarrollo normal del SNC depende del estado de la madre, de la placenta, del parto y del funcionamiento correcto del sistema cardiopulmonar del neonato. La perfusión del cerebro fetal puede verse comprometida por enfermedades sistémicas, traumatismos, intoxicaciones... de la madre, por incompatibilidades materno-fetales, por desequilibrios circulatorios en las gestaciones monocoriales, por patología de la placenta o del propio corazón fetal (1,25,29,30,42).
Las lesiones hipóxico isquémicas constituyen el trastorno mayor del periodo perinatal, con una incidencia que alcanza el 47/1000 nacimientos en algunas series (21).
Las lesiones morfológicas del SNC inmaduro difieren de las del SNC maduro, en este las lesiones morfológicas reflejan el proceso patológico que las determina y su reparación, durante la vida embrionaria tanto las anomalías genéticas como las agresiones dan lugar a malformaciones y en el periodo perinatal las lesiones neuropatológicas son determinadas por el proceso patológico, la reparación y la interferencia que estos producen en el normal desarrollo del SNC (14,21).
El estudio del SNC perinatal contribuye de forma importante al diagnóstico de la autopsia (25) y al conocimiento de una gran parte de la morbilidad neurológica infantil (21).
La mayoría de las lesiones morfológicas del SNC inmaduro no son específicas de una etiología determinada, sino el resultado de una multitud de situaciones clínicas adversas (42), el patólogo debe reconocerlas y diagnosticarlas, valorando el contexto clinicopatológico individual a la luz de la abundante literatura sobre la fisiopatología y la etiopatogenia (42).
1. VALORACIÓN DEL SNC PERINATAL
El estudio del SNC inmaduro plantea algunos problemas tanto al patólogo pediátrico como al neuropatólogo: su difícil manejo, el cambiante patrón de «normalidad» y la característica forma de reaccionar del SNC inmaduro:
a. Problemas técnicos
El SNC durante la gestación y en el periodo neonatal inmediato es blando y friable debido a su alto contenido en agua, pero normalmente tras 15 días de fijación en formol puede ser estudiado macroscópicamente realizando cortes coronales de los hemisferios y horizontales de las estructuras de fosa posterior.
La fijación durante unas horas de los bloques seleccionados para inclusión en parafina en Bouin mejora la calidad de las preparaciones al endurecer ligeramente los bloques.
b. Definición de los patrones de normalidad
El SNC durante la segunda mitad de la gestación y el primer mes de vida neonatal cambia su morfología macro y microscópica de forma bastante constante cada semana, por lo que la valoración de la existencia de alteraciones debe realizarse siempre respecto a lo esperado para una determinada edad, aunque los parámetros de normalidad hayan sido obtenidos de fetos muertos (14).
A menudo la determinación de la edad gestacional es importante en patología pediátrica. De todos los parámetros biométricos y morfológicos, los obtenidos con el examen del SNC son los más ajustados para determinar la edad gestacional, ya que el desarrollo del SNC se afecta muy poco en el crecimiento intrauterino retardado. (25,39).
Para determinar la edad gestacional debemos tener en cuenta el peso, el patrón de circunvoluciones, el tamaño del sistema ventricular, la arquitectura histológica de la corteza y el grado de mielinización (14,24,39).
Peso
El encéfalo debe pesarse en fresco, puesto que por su alto contenido en agua suele ganar peso durante la fijación, comparando el peso con el estimado para la edad gestacional (39) y comprobando que mantenga la relación adecuada con el perímetro craneal y la longitud total (14).
Una vez fijado, tras separar, por mesencéfalo, tronco y cerebelo, se pesan por separado los hemisferios y las estructuras de fosa posterior, comparando el peso de estas con el peso total. La proporción entre ambos es diferente de la del SNC maduro, ya que la tasa de crecimiento del cerebelo hasta el 5º mes de gestación es menor que la de los hemisferios (fig. 1), pero a partir de ese momento su crecimiento se acelera hasta los 18 meses, de modo que el peso del cerebelo representa un 6-7% del peso total en el último trimestre de gestación, un 9% al 4º mes de vida postnatal, un 10% al 9º y alcanza la proporción del adulto, 12%, a los 18 meses(9,24,25).
Patrón de circunvoluciones
Las circunvoluciones van apareciendo con un orden bastante constante durante la última parte de la gestación. El patrón de circunvoluciones es un parámetro fiable para establecer la edad gestacional aunque se hayan descrito algunas variaciones entre las tablas más frecuentemente utilizadas (7,9,14,24,25,39).
En la práctica conviene recordar algunos hechos más constantes: La cisura de Silvio aparece aproximadamente en la semana 16, la cisura calcarina en la 18 (fig. 1), la cisura de Rolando en la 24, el surco temporal superior en la 28, los surcos pre y postcentral en la 30, las circunvoluciones secundarias en la 32 y las terciarias a partir de la 36.
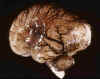
Fig. 1. RNP de 18 semanas de
edad gestacional: vista lateral del encéfalo.
Sistema ventricular
La forma y el tamaño de los ventrículos laterales cambia durante la gestación: en la semana 15 el diámetro del ventrículo lateral es dos tercios del espesor del cerebro, pero se va reduciendo conforme el cerebro crece, en la 17 es menos de la mitad, y en la 20 un tercio. El diámetro atrial permanece estable durante toda la gestación, de forma que un diámetro ventricular superior a 10 mm. en cualquier momento de la gestación hace sospechar algún tipo de patología (10,14), fundamentalmente de sustancia blanca (15,22,29,42).
El epitelio del plexo coroide fetal tiene un alto contenido en glucógeno lo que le confiere un aspecto muy claro. La cantidad de glucógeno disminuye con la maduración, que comienza en el IV ventrículo y acaba, al final de la gestación, en los ventrículos laterales (14).
Arquitectura cortical
Cerebral
Durante las primeras semanas de gestación el SNC está formado por una zona ventricular, compuesta por el neuroepitelio primitivo, a partir del cual se va a producir la emigración neuroblástica y una zona marginal (36).
Cuando los neuroblastos comienzan a emigrar hacia la superficie, empiezan a reconocerse dos capas densamente celulares, una superficial, la placa cortical y otra profunda, la zona ventricular, separadas por una zona menos celular, intermedia, constituida por las largas prolongaciones de los astrocitos radiales, que alcanzan la superficie cerebral (fig. 2) (36). La placa cortical se va a desarrollar por la emigración de los neuroblastos, guiados por las fibras radiales y, parece, atraídos por las células de Cajal Retzius (26), (aunque se desconozca a través de qué mecanismo) (36), hasta la futura primera capa, donde establecen contacto funcional con las células de Cajal Retzius y bajan a ocupar su posición definitiva en la corteza.

Fig. 2. RNP de 20 s. e. g.:
estructura histológica del lóbulo temporal: gruesa MGSEP, mejor organización
arquitectural del hipocampo que del neocórtex.
Las sucesivas oleadas de neuroblastos van a ir formando la población de la corteza , comenzando por poblar las capas profundas. La laminación comienza a ser visible cuando los neuroblastos de la capa más superficial, la 2.ª capa definitiva, han llegado a su posición final (24).
A partir de la semana 14 empieza a aparecer en la superficie pial una capa transitoria, la capa de granos superficial, que alcanza su máximo espesor en la semana 22 y comienza a desaparecer a partir de la 28, de delante atrás y de arriba abajo, de forma que pasada la semana 32 solamente queda restos en las cortezas temporal u occipital basales (9,14,24).
En la corteza de un feto de 20 semanas (fig. 2) se reconoce una capa de granos superficial (que con mucha frecuencia se desprende con la meninge) una capa molecular, (en la que podemos identificar unas neuronas de gran tamaño y disposición horizontal, las células de Cajal Retzius, neuronas transitorias, que aparecen antes de la primera oleada de migración y desaparecen al final de la gestación), y una gruesa capa neuroblástica en la que no se identifica estratificación, y cuyo limite con la c. molecular puede aparecer muy ondulante.
En la semana 24 comienza a observarse una mayor densidad celular a nivel de la 2ª capa. La progresiva maduración citológica de las neuronas va a permitir reconocer la estructura en capas en la semana 28 como áreas de distinta densidad . En la semana 32 pueden reconocerse, con HE, citoplasmas en las pirámides profundas y a partir de la 36 en ambas capas de pirámides (24,37).
Cerebelosa
La corteza cerebelosa madura de forma diferente: (9,24) mantiene una capa de granos externos hasta el final del primer año de vida; la formación de laminillas cerebelosas comienza en el vermis; los hemisferios hasta el final del 5.º mes son prácticamente lisos y el núcleo dentado presenta morfología en C, sin ondulaciones.En la semana 25 se reconoce una gruesa capa de granos externos, una fina capa molecular discretamente separada de una rudimentaria capa de granos. En la semana 32, en las laminillas de la corteza hemisférica, que todavía muestran morfología redondeada, comienzan a reconocerse neuronas de Purkinje.
Mielinización
La mielinización es un hecho capital en el desarrollo del SNC, estudiado con detalle por diferentes autores en el último siglo (7,9,14,24, 25,37).Comienza en la vida fetal y no se completa hasta el final del 2º año de vida postnatal. Tiene lugar, en sentido caudocraneal, en distintos momentos según los diferentes tractos. Existen varios detallados calendarios de mielinización (7,9,14,24,37) que pueden ser consultados para detalles de tractos específicos, siempre teniendo en cuenta la técnica con la que la mielinización ha sido valorada.
2. LESIONES CARACTERÍSTICAS DEL SNC PERINATAL
El SNC inmaduro reacciona de forma característica, aunque relativamente inespecífica, ante la falta de aporte de los elementos necesarios para su desarrollo, fundamentalmente el riego sanguíneo (12) y el oxigeno, traumatismos, toxinas o agentes infecciosos (33).
Aunque en el SNC pueden aparecer lesiones totalmente superponibles a las del SNC maduro, muchas lesiones neuropatológicas perinatales son características de este periodo y es excepcional que aparezcan el en SNC maduro (35).
La mayor parte de las lesiones que aparecen casi exclusivamente en el SNC inmaduro son debidas a accidentes hipoxico-isquémicos durante la gestación, el parto o el periodo neonatal inmediato. Pueden ser tan diferentes que incluyen desde hemorragia hasta lesiones de aspecto malformativo por interferencia con el desarrollo normal (21,25), y con frecuencia se presentan asociadas (15,29,42).
a. Hemorragia
La hemorragia es la lesión hipóxico-isquémica (HI) más frecuente del SNC inmaduro (14) probablemente debido a que las células endoteliales del SNC, por su contenido en mitocondrias, 3-5 veces mayor que el del resto de los capilares del organismo (42), tienen altos requerimientos de metabolismo oxidativo y sufren necrosis en los estados asfícticos (35).
La distribución de las hemorragias es prácticamente superponible en los fetos muertos anteparto y en los recién nacidos muertos (14,29).
Las lesiones hemorrágicas HI más características del SNC inmaduro son:
Hemorragia en matriz germinal subependimaria (HMGSEP)-hemorragia intraventricular (HIV)
La Hemorragia intraventricular (HIV) es la hemorragia intracraneal más frecuente en el neonato y es característica del Recién Nacido Pretérmino (RNP) (42). Su incidencia varía según las series entre el 26 y el 80%. Aunque su incidencia ha bajado en los últimos años, el aumento de la supervivencia de RNP menores de 1000g indica que va a continuar siendo un problema en neonatología (21,42).
Es una lesión preferentemente postnatal (42), sucede más frecuentemente en las primeras 72 horas de vida, pero puede producirse intraútero (1,6,14,24,29,35,39).
La mayoría de las HMGSEP aparecen en la zona de matriz que persiste hasta el final de la gestación, la localizada en el surco caudado-talámico, a nivel del agujero de Monro (fig. 3). En el RNP menor de 30 semanas de edad gestacional puede aparecer en cualquier zona de los ventrículos laterales, incluso en el techo del IV ventrículo.
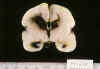
Fig. 3. RNP de 25 s. e. g. y 5
días de vida: hemorragia bilateral en MGSEP, HIV con dilatación del
ventrículo lateral izquierdo.
La MGSEP es un acúmulo de neuroblastos y espogioblastos programados para emigrar, situado en torno a las paredes de los ventrículos laterales, con vasos de diámetro luminal relativamente grande y pared constituida exclusivamente por una única capa de células endoteliales. La necrosis de las células endoteliales determina la extravasación hemática, que difunde rápidamente, debido a la ausencia de estroma y a que los neuroblastos poseen actividad fibrinolítica (14,42), rompe el revestimiento ependimario y se abre a la luz ventricular, con mayor frecuencia en los RNP menores de 1.500 g. Una vez en el sistema ventricular (fig. 3), la sangre es drenada con el LCR, ocupando sucesivamente el III y IV ventrículos, las cisternas basales y ambos valles silvianos (fig. 4).
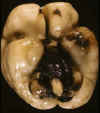
Fig. 4. RNP de 25 s. e. g. y 3
días: hemorragia subaracnoidea en fosa posterior y cisternas basales, drenando
HIV.
En algunas ocasiones la extravasación hemática se extiende además al parénquima adyacente, fundamentalmente a la sustancia blanca del centro oval o al n. caudado. Estas zonas de hemorragia intraparenquimatosa asociadas a HIV pueden ser muy difíciles de distinguir de áreas de necrosis hemorrágica de la sustancia blanca, ya que incluso microscópicamente, lo único que es posible observar es material hemático sustituyendo las estructuras y no suelen identificarse áreas de necrosis en las zonas periféricas.
Puesto que la severidad de la HIV inicial parece ser crítica para el pronóstico (42), la HIV del pretérmino ha sido clásicamente dividida, desde el punto de vista de la neuroimagen, fundamentalmente ecográfico, en cuatro grados:
Grado I: Hemorragia localizada en la MGSEP.
Grado II: HIV sin dilatación ventricular.
Grado III: HIV con dilatación ventricular.
Grado IV: HIV con extensión parenquimatosa.
Volpe recientemente (42) ha dejado de considerar el grado IV, al interpretar que las lesiones hemorrágicas del parénquima que acompañan a las HIV de mayor volumen no son extensiones parenquimatosa sino infartos hemorrágicos de la sustancia blanca.
La HIV del RNP evoluciona con mucha rapidez. En muy poco tiempo el sistema ventricular puede encontrarse libre de contenido hemático, excepto en los cuernos occipitales donde persiste algo más por el decúbito. En estos casos, el examen externo revela la característica Hemorragia Subaracnoidea en la fosa posterior y ambos valles silvianos (fig. 4). En los cortes coronales puede apreciarse asimetría ventricular y en la zona donde se produjo la HMGSEP una cavidad o hendidura, cuyo estudio histológico mostrará uno o varios quistes de tamaño variable (fig. 5), con escasa respuesta glial y macrofágica, rodeados de células germinales. En la superficie ventricular puede observarse gliosis subependimaria, a veces con prominente formación de rosetas, incluso de septos, siderófagos en la luz (fig. 6) o entremezclados con la gliosis y/o excrecencias de matriz germinal. El epitelio del plexo coroide puede mostrar depósito de pigmento férrico.
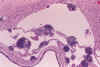
Fig. 5. RNP fallecido 20 días tras
HIV: quistes en MGSEP, separados por tabiques gliales, con restos de
neuroblastos y escasa respuesta macrofágica.
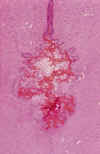
Fig. 6. Obstrucción completa del
acueducto de Silvio por gliosis, siderófagos y hematíes en RNP con
hidrocefalia posthemorrágica.
La reabsorción del material hemático se hace fundamentalmente en el espacio subaracnoideo, cuyos macrófagos muestran primero una intensa eritrofagocitosis y posteriormente acúmulo de pigmento férrico. Al mismo tiempo se desencadena proliferación de fibroblastos. La siderosis y fibrosis meníngea, pueden obstruir los orificios de salida del IV ventrículo, la causa más frecuente de hidrocefalia posthemorrágica, junto a la obstrucción, por gliosis, del acueducto de Silvio (fig. 6). La ocupación del espacio subaracnoideo de la fosa posterior, al interferir con la nutrición de la capa de granos externos del cerebelo, puede ser la causante de la frecuente asociación de atrofia focal de laminillas en el cerebelo, que a veces puede llegar a ser muy extensa.
La extensión parenquimatosa evoluciona rápidamente mediante licuefacción y cuando interesa la cápsula interna puede acompañarse de degeneración secundaria del haz piramidal.
Hemorragia subaracnoidea
La hemorragia subaracnoidea es una lesión muy frecuente del SNC, tanto del RNT (3) como del RNP (29), muerto ante o postparto, con la que se han asociado múltiples factores de riesgo (14). Puede ser microscópica, focal o difusa.
Hemorragia subpial
La inmensa mayoría de las hemorragias subpiales son microscópicas, pero pueden coalescer formando hematomas subpiales fundamentalmente en el lóbulo temporal y en el cerebelo.
Hemorragia cerebelosa
Es una lesión característica del RNP, mucho menos frecuente que la HIV (29). Aunque se ha puesto en relación con factores de riesgo muy concretos (42), la mayoría de las hemorragias cerebelosas se forman por coalescencia de las microhemorragias subpiales, probablemente debido a que los granos externos del cerebelo poseen, como las células de la matriz germinal, actividad fibrinolítica.
Hemorragia del plexo coroide
El plexo coroide puede ser el origen de HIV, tanto en el RNP como, más frecuentemente, en el RNT (3,42). En el primero puede coexistir con HMGSEP y en el segundo ser la única fuente del sangrado. Se comprueba por la demostración histológica de hemorragia en la estroma del plexo.
b. Edema cerebral
La capacidad del SNC inmaduro para desarrollar edema ante insultos hipoxico-isquémicos continúa debatido en la literatura (14,34,42) aunque existe acuerdo en que la herniación prácticamente no existe (14,34,42), probablemente en relación con la distensibilidad de las fontanelas.
Considerando como criterios para diagnosticar edema, un peso mayor del esperado para la edad, con un aumento de peso tras la fijación mayor del habitual en los cerebros perinatales, aplanamiento de circunvoluciones, en los cerebros a término, y colapso ventricular (fig. 7), en nuestra experiencia es la lesión más frecuentemente observada en los neonatos y fetos fallecidos muy inmediatamente tras el episodio agudo (27,29), habitualmente asociado a microhemorragias, como única lesión objetiva. En estos casos el SNC es aún más friable y blando de lo habitual y en los cortes seriados suele mostrar una inadecuada fijación de las estructuras profundas, que no mejora prolongando el periodo de fijación.

Fig. 7. RNP de 36 s. e. g.:
aplanamiento de circunvoluciones y colapso ventricular (presencia de bario por
arteriografía postmortem).
c. Lesiones de la sustancia blanca
La sustancia blanca (SB) del SNC inmaduro es especialmente vulnerable (1,14,15,16,33,34, 35,38,42), tanto en la vida intrauterina como en la postnatal (1,29,42).
Las lesiones de la SB incluyen: necrosis, gliosis y trastornos de mielinización, pueden aparecer independientes o, más frecuentemente, asociadas y han recibido nombres tan diversos como leucomalacia periventricular, leucoencefalopatía telencefálica perinatal y leucomalacia subcortical.
Necrosis
La necrosis de la SB, originalmente denominada leucomalacia periventricular, es una lesión característica del RNP, generalmente postnatal (42). Aunque se ha considerado el principal sustrato morfológico de las secuelas neurológicas de los RNP (15,20,22,31,40,42) es menos frecuente que la gliosis en las series neuropatológicas (1,15,29).
Características macroscópicas
La necrosis aguda de la SB puede aparecer como un área mal definida de reblandecimiento translucido con o sin hemorragia o como manchas blancas o blanco-amarillentas de 2-6 milímetros de diámetro, generalmente localizadas en torno al ángulo lateral de los ventrículos laterales (7,9,25,33,34,35).
En la fase crónica la SB dañada puede aparecer retraída, reducida de volumen, con o sin estrías blanquecinas o herrumbrosas o bien quística (fig. 8) con quistes de tamaño y distribución variable, uni o multiloculados. Los ventrículos suelen estar dilatados, con frecuencia de forma asimétrica y el cuerpo calloso adelgazado (22,30,31,32).
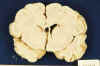
Fig. 8. RNP de 22 días: asimetría
ventricular. Quiste junto al ángulo del VLD.
Características microscópicas
Las lesiones agudas se caracterizan por áreas de necrosis coagulativa, intensamente eosinófilas (7,9,14,33,34) (fig. 9). Hacia las 12 horas comienza a observarse proliferación microglial, cuyo papel en la patogenia de las lesiones de la SB ha sido recientemente resaltado (40). Entre las 12 y 48 pueden apreciarse bolas axonales, pero es excepcional observar leucocitos. Posteriormente en las fibras lesionadas con frecuencia se deposita material basófilo, que les hace semejantes a hifas (33) (fig. 10).

Fig. 9. Necrosis aguda de SB:
intensa eosinofilia de las fibras.
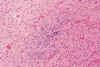
Fig. 10. Necrosis evolucionada de
SB: depósito de material basófilo en las fibras necróticas, macrófagos y
gliosis.
Cuando la lesión es difusa la lesión más frecuentemente observada es microespongiosis, preferentemente de las zonas profundas, asociada a cariorrexis de la glia (14).
En las fases crónicas las lesiones se delimitan mejor por la presencia de macrófagos (fig. 10), que pueden mostrar citoplasma esmerilado («Gitter cells») o cargado de pigmento férrico y gliosis reactiva (14,33). En los espacios de Virchow-Robin adyacentes suelen observarse macrófagos. Ocasionalmente pueden apreciarse hendiduras de colesterol y reacción granulomatosa de cuerpo extraño (33). Las lesiones quísticas muestran proliferación gliovascular y macrófagos en sus bordes (33,34).
Gliosis
La gliosis de la S.B es la lesión de S.B. más frecuentemente observada en las series neuropatológicas (1,15,29) y puede ser por tanto uno de los sustratos morfológicos de las secuelas neurológicas de los RN sin lesiones ecográficas en el periodo neonatal (16,29, 28,38). Puede observarse en el RNP y en el RNT así como en fetos muertos anteparto con infartos de placenta (1,29) o de gestaciones múltiples (30).
Características macroscópicas
No existen características macroscópicas distintivas de la gliosis de sustancia blanca. En la fase aguda puede apreciarse colapso ventricular con un aspecto gelatinoso translucido de la sustancia blanca (fig. 11).
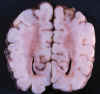
Fig. 11. RNP de 32 s. e. g. y 40
días: coloración parda translúcida de SB occipital.
En las fases crónicas, por el contrario el hallazgo macroscópico más frecuente es la reducción de volumen de la SB, con adelgazamiento del cuerpo calloso y dilatación ventricular (fig. 14) (14,22,33,34).
Características microscópicas
El diagnóstico histológico se basa en la proliferación numérica de los astrocitos, que puede ser subjetiva, junto a binucleación o signos de hipertrofia astrocitaria, que en la SB suele manifestarse por tres tipos de modificaciones citoplásmicas: (33,34,35) (fig. 12).
Presencia de citoplasma estrellado con finas prolongaciones.
Aspecto gemistocítico con un citoplasma redondeado eosinófilo, aproximadamente del tamaño del núcleo del que salen prolongaciones cortas.
Presencia en el citoplasma de los astrocitos de aspecto gemistocítico de una zona eosinófila, bien delimitada, rodeada a veces por un halo claro (que con ME corresponden a acúmulos de gliofibrillas).
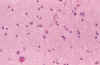
Fig. 12. Gliosis de SB: astrocitos
hipertróficos de diferente morfología y espongiosis.
Junto a la proliferación astrocitaria en las primeras fases suele apreciarse espongiosis del neuropilo (fig. 12).
Con frecuencia la gliosis se acompaña de la presencia de pequeños acúmulos globulosos de material anfófilo (14), tanto aisladas en la SB como en localizacióm perivascular, que aunque no siempre se tiñen con el Von Kossa, parecen microcalcificaciones (29). En ocasiones, fundamentalmente en cardiopatías congénitas en nuestra experiencia, se asocia con heterotopia de neuronas maduras aisladas en la SB (27), quizas por interferencia con la función de guía de las fibras radiales.
En las fases más crónicas se suele acompañar de disminución de la mielinización.
d. Lesiones de la sustancia gris
Mientras la mayoría de las lesiones hemorrágicas y de la sustancia blanca son casi exclusivas del SNC inmaduro, las lesiones de la sustancia gris son más parecidas a las del niño mayor o el adulto.
En el SNC inmaduro se pueden observar infartos en los territorios de las diferentes arterias cerebrales, necrosis cortical en los territorios limítrofes, necrosis cortical laminar, totalmente superponibles a las del SNC maduro con la única diferencia que la licuefacción suele tener lugar más precozmente que en el SNC maduro (9,34,35,42).
Algunas lesiones son muy características y casi exclusivas del SNC perinatal:
Características macroscópicas
En los casos fallecidos pocas horas tras un episodio HI el único hallazgo suele ser el edema cerebral.
El aspecto macroscópico de encefalopatía HI en el periodo perinatal consiste con frecuencia en una coloración marrón-rojiza difusa de la sustancia blanca, contrastando con la palidez del ribete cortical, el llamado «ribbon-effect» (14,213334, 35) (fig. 13).
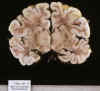
Fig. 13. Encefalopatía hipósico
isquémica en RNT con hipoplasia de ventrículo izquierdo: intensa congestión
de SB, contrastando con la palidez cortical.
El tejido necrótico puede presentar un llamativo color amarillo-verdoso en los neonatos ictéricos, pero en general, las lesiones agudas focales suelen ser difíciles de reconocer macroscópicamente (34).
La necrosis subaguda de la s. gris se caracteriza por cambios de coloración, con reblandecimiento, que hace que se separe de la s. blanca (34).
Las lesiones necróticas evolucionadas suelen ser quísticas (fig. 14), pero pueden aparecer como áreas de mayor consistencia intensamente blancas, fundamentalmente en el tálamo, los núcleos grises de la base y el tronco (34,35).
En los niños que mueren meses o años tras una lesión HI difusa el cerebro pesa mucho menos de lo normal, existe dilatación ventricular y en los cortes seriados se pueden apreciar áreas quísticas y/o escleróticas, junto a zonas de atrofia cortical, que cuando afectan a las zonas profundas de los surcos con respeto de las crestas recibe el nombre de ulegiria (fig. 14) (9,34,39,42). A menudo la afectación del cerebro es mucho más intensa que la del cerebelo, lo que determina una neta desproporción entre el tamaño de ambos (34,35).
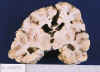
Fig. 14. Dilatación ventricular,
atrofias corticales focales bilaterales, con patrón ulegírico, y quistes en
estriado en niña de 2 años con encefalopatía postanoxia de parto.
Características microscópicas
Necrosis neuronal
La necrosis neuronal selectiva es la lesión más frecuente en la encefalopatía HI neonatal (42). Aunque se ha considerado una lesión del RNT, últimamente se está resaltando en la literatura la importancia de la necrosis neuronal en el RNP (16,28,29), como posible sustrato de déficit neurológico en RNP sin lesiones parenquimatosas ecográficas en el periodo neonatal.
Frecuentemente se asocia a otras lesiones (28,33,42).
El aspecto histológico de la necrosis neuronal depende del grado de maduración de las neuronas y del tiempo transcurrido hasta el examen histológico.
El primer cambio, apreciable tras una hora, es la vacuolización citoplásmica causada por hinchazón mitocondrial (42). Si han transcurrido 24-36 horas, las neuronas maduras muestran eosinofilia citoplásmica y picnosis nuclear, mientras que en las inmaduras el único criterio fiable es la presencia de cariorrexis (fig. 15) (14,33,34,35). Simultáneamente empieza a observarse activación microglial (42) en forma de microglia en bastoncillo.
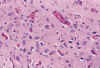
Fig. 15. Cariorrexis y eosinofilia
neuronal en núcleos propios de protuberancia.
La topografía y la extensión depende de la severidad y duración del insulto y de la edad gestacional (42).
La necrosis neuronal de los núcleos propios de protuberancia en el neonato, fue descrita por Adams en 1967. Unos años más tarde Friede describió la necrosis neuronal del subiculum asociada a necrosis pontina, acuñando el término de «necrosis ponto-subicular» para definir un tipo especial de encefalopatía neonatal. (9,35,42). No existe acuerdo en cuanto a la constancia de esta asociación (35). En nuestra experiencia (27,29) los núcleos propios de protuberancia y el subiculum son las dos zonas del SNC neonatal donde con mayor frecuencia puede reconocerse necrosis neuronal, tanto en el RNP como en el RNT, pero raramente se asocian: la cariorrexis de las neuronas de la porción anterior de protuberancia (fig. 15) es la lesión histológica más frecuente en los cerebros de los fetos muertos anteparto tras un episodio agudo , mientras que en el RN es más frecuente la necrosis neuronal en el subiculum, quizás porque en los primeros con frecuencia la región del hipocampo no se encuentra en condiciones de ser estudiada histológicamente.
La necrosis neuronal en el tálamo es muy frecuente en el SNC perinatal (14,33,34,35), donde la necrosis neuronal aguda se suele expresar por eosinofilia citoplásmica y picnosis neuronal. Las neuronas necróticas en el tálamo tienden a permanecer, con un citoplasma intensamente basófilo, PAS+, que posteriormente se incrusta de cantidades variables de calcio y hierro (fig. 16). Las lesiones crónicas del tálamo y del troncoencefalo en el periodo perinatal se caracterizan histológicamente por la presencia de neuronas incrustadas (momificadas), gliosis, algunos macrófagos y un grado variable de microcalcificaciones intraparenquimatosas en forma de pequeños acúmulos globulosos hematoxinófilos perivasculares o de incrustación de prolongaciones del neuropilo, que pueden llegar a hacerse confluentes (4,33,34), fundamentalmente en el tronco. Algunas veces se acompañan de calcificaciones parietales vasculares en arterias de pequeño tamaño (fig. 17) intraparenquimatosas o meníngeas. La calcificación de las arterias del putamen que puede ser detectable por ecografía (2) han recibido el nombre de vasculopatia lenticuloestriada, y se considera un marcador de daño antenatal (1,33,34,35). Es excepcional observar en el periodo neonatal hipermielinización del tálamo, «Status marmoratus», (34,35,42).

Fig. 16. Gliosis,
microcalcificaciones e incrustación neuronal en el tálamo de RNP con crisis
desde el nacimiento.
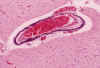
Fig. 17. Calcificaciones parietales
en una arteria del putamen del mismo caso que la fig. 16.
Gliosis
El aspecto histológico de la respuesta astrocitaria depende en parte de la localización:
En la corteza la proliferación astrocitaria suele expresar hipertrofia nuclear, adoptando la morfología astrocitos con núcleos muy claros desnudos, semejante a la gliosis de Alzheimer tipo II del SNC maduro, pero sin relación con la disfunción hepática en el neonato (33,34).
La gliosis en el tálamo y núcleos grises subcorticales suele estar constituida por astrocitos, probablemente protoplásmicos, a los que no se les reconoce citoplasma con la HE (33,34) (fig. 16).
La proliferación glial a nivel de la capa de neuronas de Purkinje, no adopta la morfología de la gliosis de Bergmann del SNC maduro, sino que suelen ser astrocitos uni o bipolares (33,34).
En los núcleos propios de protuberancia y en la oliva suelen mostrar citoplasma estrellado visible con la HE (33,34).
e. Encefalopatías quísticas perinatales
Las lesiones destructivas quísticas severas del SNC inmaduro, que semejan malformaciones, han recibido múltiples nombres y se han relacionado con diferentes etiologías (7,8,9,14, 24,25,33,34,35,39,42).
Pueden dividirse en dos grandes grupos: las encefalopatías quísticas vasculares fetales (hidranencefalia y porencefalia) y la encefalopatía multiquística (21,25,35,39).
Encefalopatías quísticas vasculares fetales
Son lesiones disruptivas quísticas secundarias a accidentes vasculares durante la vida intrauterina.
Hidranencefalia
Es la ausencia prácticamente completa de la convexidad de los hemisferios cerebrales, que se encuentran sustituidos por cavidades recubiertas por meninge y ocupadas por LCR. La calota está normal o distendida. La cara basal de los lóbulos occipitales y temporales, el tálamo, la porción posterior de los núcleos de la base, el tronco y el cerebelo se encuentran conservados generalmente (14,21,25,34,42).
Se considera que está causada por un accidente cerebrovascular intraútero en el territorio de ambas carótidas (9,14,25,34,39).
Porencefalia
Es la presencia de una cavidad que afecta a todo el espesor del parénquima, comunicando el sistema ventricular con la superficie meníngea. Puede ser uni o bilateral y se localiza en algún territorio arterial, más frecuentemente en el de la arteria cerebral media (fig. 18). Se consideran lesiones necróticas, por interrupción del flujo arterial durante la vida fetal. El parénquima circundante a la lesión quística suele presentar diferentes trastornos de emigración, que reflejan el momento de aparición de la lesión disruptiva (9,14,25,34,39).

Fig. 18. RNP de 28 s. e. g. y 4
días: porencefalia bilateral en el territorio de las arterias cerebrales
medias.
Encefalopatía multiquística
Es la sustitución de diferentes áreas del cerebro por cavidades de diferentes tamaños, que afectan fundamentalmente a la corteza y a la sustancia blanca (fig. 19). Es el resultado de un severo insulto hipóxico-isquémico o infeccioso en el último trimestre de la gestación o en el periodo neonatal (8,9,14,33,34,39,42).
Fig. 19. RNT de 38 s. e. g. y 2
horas, madre adicta a drogas por vía parenteral: encefalopatía multiquística.
f. El SNC en el síndrome de transfusión feto-fetal
Los RN de gestación múltiple, fundamentalmente los gemelos monocoriales, muestran con mayor frecuencia que los RN de embarazo único morbilidad neurológica que no siempre es achacable a la prematuridad o al bajo peso (5,17,30,31).
En el síndrome de transfusión feto-fetal, el gemelo donante y el receptor tienen un riesgo significativo de presentar un amplio espectro de lesiones en el SNC (17,25,30,31).
La morfología de las lesiones depende de la severidad y tipo de trastorno hemodinámico implicado, el momento de la gestación en que sucede y el tiempo transcurrido entre el accidente y el estudio del SNC (25). En el gemelo donante las lesiones suelen ser hipóxico-isquémicas, debido a la hipovolemia y anemia crónicas y en el receptor hemorrágicas (21,25,30,31).
Las lesiones más graves, se observan en el SNC de aquellos gemelos que han sobrevivido a la muerte intraútero de su gemelo (fig. 20) (21,25,32,41,42). Cuando un gemelo donante muere, el receptor sufre una brusca deprivación de flujo sanguíneo, al derivar súbitamente la sangre al sistema vascular sin tensión arterial del donante muerto, debido a la íntima relación de las circulaciones fetales en las gestaciones monocoriales (21,25,32,41,42). Las lesiones encefálicas del gemelo superviviente suelen ser difusas y generalmente consisten en micrencefalia con diversas lesiones destructivas (fig. 20) quísticas o no (21,25,32). Con menor frecuencia se han descrito lesiones en territorios vasculares (hidranencefalia, porencefalia) en cuya patogenia se ha implicado el paso de restos necróticos y/o sustancias tromboplásticas desde el gemelo muerto a la circulación del superviviente (25,42).

Fig. 20. RNP de 28 s. e. g.,
embarazo monocorial, gemelo muerto 5 semanas antes del parto: micrencefalia con
quistes preferentemente corticales y marcada dilatación ventricular.
g. Retraso en el desarrollo
Es un tipo de «lesión» única del SNC perinatal. Puede expresarse como un fallo global de crecimiento, que se expresa normalmente por micrencefalia con microcefalia, o como un fallo en adquirir un nivel de desarrollo adecuado a la edad (14). En nuestra experiencia el más frecuente es el desarrollo insuficiente de los opérculos (27), puede asociarse a otras lesiones, fundamentalmente a gliosis de la sustancia blanca.
h. Lesiones de la médula espinal
La mayoría de las lesiones adquiridas de la médula espinal en el neonato son consecuencia de trauma o asfixia en el momento del parto.
Las lesiones asfictícas son superponibles a las del encéfalo (hemorragia, necrosis neuronal) aunque mucho menos frecuentes (33).
Las lesiones traumáticas de la médula en la actualidad, afortunadamente, son poco frecuentes, pero pueden aparecer como complicación de los partos de nalgas, en cefálica con forceps o en los que se produce una hiperextensión del cuello (13,21,34,42). En los partos de nalgas la lesión suele localizarse en la médula cervical baja o torácica alta y el mecanismo suele ser una tracción excesiva, mientras que en los partos en cefálica con fórceps la lesión suele suceder en la unión bulbo-medular o médula cervical alta y el mecanismo por torsión junto a hiperextensión (13,21,42). En ambos casos se puede producir necrosis medular, que puede llegar a afectar a toda la sección de la médula junto a hemorragias tanto intra como extradurales. La organización de la hemorragia epidural puede acompañarse de lesiones vasculares diferidas (13).
En los casos más graves la lesión medular puede acompañarse de fracturas y/o luxaciones vertebrales.
BIBLIOGRAFÍA
Burke CJ, Tannenberg AE. Prenatal brain damage and placental infarction. An autopsy study. Developmental Medicine and Child Neurology 1995; 37: 555-62.
Cabañas F, Pellicer A, Morales C, García-Alix A, Stiris T. New pattern of hyperechogenicity in thalamus and basal ganglia studied by Doppler flow imaging. Pediatr Neurol 1994; 10: 109-16.
Campos-Castelló J, de Santos MT, Jiménez-Álamo ML, Ramírez R, Careaga-Maldonado ML. Hemorragias cerebrales en el recién nacido a término. Rev Neurol 1999; 29: 239-46.
Cusí V, Iriondo M, Campistol J, Krauel J. Síndrome de Moebius asociado a ingesta de misoprostol durante la gestación. Neurología 2000; 15: 586 (abstract).
Denbow ML, Battin MR, Cowan F, Azzopardi D, Edwards AD, Fisk NM. Neonatal cranial ultrasonographic findings in preterm twins complicated by severe fetofetal transfusion syndrome. Am J Obstet Gynecol 1998; 178: 479-83.
De Vries LS, Eken P, Groenendaal F, Rademaker KJ, Hoogervorst B, Bruinse HW. Antenatal onset of haemorrhagic and/or ischemic lesions in preterm infants: prevalence and associated obstetric variables. Arch Dis Child Fetal Neonatal Ed. 1998; 78: F51-F56.
Esiri MM, Oppenheimer DR. Neuropathology of the very young, cp. 21 en Diagnostic Neuropathology, pp. 320-332, Blackwell Sci. publ. Oxford, 1999.
Ferrer I, Navarro C. Multicystic encephalomalacia of infancy: Clinicopathological report of 7 cases. J Neurol Sci 1978; 38: 179-89.
Friede RL. Developmental Neuropathology, 2.ª ed. Springer-Verlag: Berlin, 1989.
García Alix A, Cabañas F, Morales C, Pellicer A, Echevarria J, Paisan L, Quero J. Cerebral abnormalities in congenital myotonic dystrophy. Pediatr Neurol 1991; 7: 28-32.
García Alix A, Morales C, Martín-Ancel A, Cabañas F, Pellicer A, Quero J. Prognostic value of CSF Neuron Specific Enolase (NSE) in comatose term infants with severe hypoxic-ischemic brain injury. Pediatr Research 1995; 38: 3, Abstract 66.
García Alix A, Stiris TA, Morales C, Hernanz y Valcarce M. Newborn piglet models to study perinatal brain damage. Pediatr Research 1995; 38: 3, Abstract 67.
García Gonzalez PL, García Alix A, García Rubio B, Morales C, Cabañas F, Pellicer A, Quero J. Lesión medular cervical en le recién nacido. An Esp Pediatr 1992; 37: 419-21.
Gilles FH. Perinatal Neuropathology: Textbook of Neuropathology, cap. 9, 3.ª ed., Davis R.L. y Robertson, D.M. (Eds.), pp. 331-185, Williams & Wilkins: Baltimore, 1997.
Gilles FH, Levinton A, Golden JA, Paneth N, Rudelli RD. Groups of histopathologic abnormalities in brains of very low birthweight infants. J Neuropathol Exp Neurol 1998; 57: 1026-34.
Golden J. Neonatal white matter disease: new thoughts on an old problem. Brain Pathol 2000; 10: 791 (abstract).
Grether JK, Nelson KB, Cummins SK. Twinning and cerebral palsy: experience in four northern California counties, births 1983 trough 1985. Pediatrics 1993; 92: 854-858.
Hirayama A, Okoshi Y, Hachiya Y, Ozawa Y, Ito M, Takashima S. Early immunohistochemical detection of axonal damage and glial activation in extremely immature brains with periventricular leukomalacia. Brain Pathol 2000; 10: 791 (abstract).
Iida K, Takashima S, Takeuchi Y, Ueda K. Neuropathologic study of newborns with prenatal-onset leukomalacia. Pediatr Neurol 1993; 9: 45-8.
Inder T, Huppi PS, Zientara GP, Maier SE, Jolesz FA, Di Salvo D, Robertson R, Barnes PD, Volpe J. Early detection of periventricular leukomalacia by diffusion-weighted magnetic resonance imaging techniques. J Pediatr 1999; 134: 631-4.
Kinney HC, Armstrong DD. Perinatal Neuropathology, en Greenfield’s Neuropathology, 6.ª ed., Graham D.I. y Lantos P.L. (Eds.) 1997, vol.1, pp. 537-600.
Kuban K, Sanocka U, Leviton A, Allred EN, Pagano M, Dammann O, Share J, Rosenfeld D, Abiri M, Di Salvo D, Doubilet P, Kairam R, Kazam E, Kirpekar M, Schonfeld S. White matter disorders of prematurity: association with intraventricular hemorrhage and ventriculomegaly. J Pediatr 1999; 134: 539-46.
Kuban KCK. Preterm white matter disease and inflamation/infection. Brain Pathol 2000; 10: 790 (abstract).
Larroche JC, Encha-Razavi F. The Central Nervous System, cap 21 en Textbook of Fetal and Perinatal Pathology Wigglesworth J.S y Singer D.B. (Eds) Blackwell Sci. Publ., Oxford, 1991.
Larroche JC, Encha-Razavi F, De Vries L. Central Nervous System. En Potter’s Pathology of the Fetus and Infant, Gilbert-Barness E. (Ed) vol. 2, pp 1028-1150, Mosby-Year Book, Inc. St. Louis Missouri, 1997.
Marín-Padilla M. Desarrollo de la corteza cerebral humana. Teoría arquitectónica. Rev Neurol 1998; 29: 208-16.
Ortega L, Morales C, de la Rosa P, Hernández R, Valverde E, Cabañas F. Lesiones neuropatológicas asociadas a cardiopatía congénita. Neurología 1996; 11: 448 (abstract).
Paneth N. Classifying brain damage in preterm infants. J Pediatr 1999; 134: 527-9 (Editorial).
Pérez Mies B, Morales C, Esteban I, Pérez Alonso P, Martinez M, Rodríguez JI. Neuropatología del recién nacido pretérmino: revisión de 417 autopsias. Neurología 1999; 14: 142-143 (abstract).
Pérez Mies B, Morales C, Esteban I, Rodríguez JI. Neuropatología del recién nacido pretérmino en las gestaciones múltiples: revisión de 74 autopsias. Neurología 2000; 15: 588 (abstract).
Perlam JM, Broyles RS, Rogers CG. Neonatal neurologic characteristics of preterm twin infants <1,250 gm. Birth weight. Pediatr Neurol 1997; 17: 322-326.
Pharoah POD. Consequences of in-utero death in a twin pregnancy. Lancet 2000; 355: 1597-602.
Rorke LB. Pathology of perinatal Brain Injury, Raven Press: New York, 1982.
Rorke LB. Perinatal brain damage. Greenfield’s Neuropathology, 5.ª ed, Adams J.H. y Duchen L.W. (Eds), pp. 639-709, Edward Arnold: London, 1992.
Rorke LB. Anatomical features of the developing brain implicated in pathogenesis of hypoxic-ischemic injury. Brain Pathol 1992; 2: 211-221.
Sarnat HB. La migración neuroblástica: aspectos embriológicos y mecanismos. Rev Neurol 1998; 27: 242-246.
Smith JF. Pediatric Neuropathology, McGraw-Hill, New York, 1974.
Stanley F. Epidemiology of neonatal white matter disease. Brain Pathol 2000; 10 : 790 (abstract).
Stocker JT, Dehner LP. Pediatric Pathology, J.B. Lippincott comp: Philadelphia, 1992.
Tahraoui SL, Marret S, Bodénant C, Leroux P, Dommergues MA, Evrard P, Gressens P. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter Brain Pathol 2001; 11: 56-71.
Van Heteren CF, Nijhuis JG, Semmenkrot BA, Mulders LGM, Van den Berg PP. Risk for surviving twin after fetal death of co-twin in twin-twin transfusion syndrome. Obstet Gynecol 1998; 92: 215-9.
Volpe JJ. Neurology of the Newborn, 4.ª ed W.B. Saunders, Philadelphia, 2001.
![]()