
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
Francisco Mampaso1, Elena Nieto1, Guillermo Pérez de Lema2
1 Hospital Universitario Ramón y Cajal. Universidad de Alcalá. Madrid. 2 Medizinische Poliklinik, Ludwig-Maximilians Universität, Munich.
El daño inmunológico renal en humanos se clasifica habitualmente de acuerdo con el conocimiento de los mecanismos que aporta el estudio de diversos modelos de experimentación. La extrapolación clínica de estos mecanismos no se corresponde con el nivel de conocimiento que aportan dichos modelos, en parte, debido a las limitaciones que conlleva el estudio del tejido renal en el momento de su obtención, generalmente una vez que la enfermedad está ya establecida. Este hecho hace complejo conocer que mecanismos inician el daño como que otros son responsables de su recuperación y/o su progresión. La mayoría de los diagnósticos clínicos se sustentan en la obtención de una muestra de tejido renal que sirve para establecer una clasificación morfológica utilizando la microscopia óptica y el estudio ultraestructural, así como para caracterizar el tipo de depósito inmune presente mediante técnicas de inmunofluorescencia y/o inmunohistoquímica. Otros estudios complementarios incluyen la caracterización del fenotipo del componente inflamatorio celular existente y/o la presencia de mediadores inflamatorios a nivel de la expresión de la proteina o del ARNm (1-3).
En este trabajo se expondrán aquellos modelos experimentales que han permitido conocer el papel que juegan derterminados antígenos (Ags) en la respuesta inmune responsable del inicio del daño renal (tabla I). Los Ags pueden formar parte de la propia estructura renal como los que constituyen la membrana basal glomerular (MBG) o estar localizados en la superfice de las células intrínsecas del glomérulo renal. Alternativamente, los Ags responsables de la formación y depósito de complejos inmunes (CI) nefritogénicos pueden tener un origen extraglomerular, ya sea a través de CI preformados en la circulación con posterior acúmulo en el glomérulo o mediante atrapamiento y depósito en el glomérulo del Ag, reacción con el Ac y formación in situ de CI. Aunque la respuesta inmune es habitualmente humoral —mediada por anticuerpos (Acs)—, existen situaciones de daño renal en las que la respuesta inmune nefritogénica es predominantemente celular (4).
Acs QUE REACCIONAN CON Ags GLOMERULARES
Ags de la MBG
Enfermedad por Acs anti-MBG
A. Patología en Humanos. La enfermedad por Acs anti-MBG es el ejemplo más representativo de daño glomerular en el que el Ac reacciona con la MBG. Además, el Ac reacciona con la membrana basal alveolar (MBA) y da lugar a una hemorragia pulmonar (síndrome de Goodpasture), en aproximadamente un 50% de los pacientes. El papel de los Acs anti-MBG en el daño glomerular fue confirmado en 1967, al demostrar el Ac depositado en la MBG con un patrón lineal, similar al que se observa en su correspondiente modelo experimental (5). El Ag nefritogénico ha sido identificado como la porción globular del domino no colágeno (NC1) de la cadena a3 del colágeno tipo IV (fig. 1a) (6).

Fig. 1. A) Enfermedad de
Goodpasture. Depósitos lineales de IgG (IF x 550); B) Fase heteróloga en el
modelo de nefritis nefrotóxica. Acúmulo de neutrófilos (PAS x 300); C) Fase
autóloga con infiltracioón por células ED1+(FAAFA x 550) y, D) Acs anti-MBG
en el modelo de nefritis nefrotóxica (IF x 250).
B. Modelos Experimentales. El modelo homólogo de la enfermedad por Acs anti-MBG en humanos, ha sido estudiado en sus dos formas pasiva y activa. La forma pasiva de la enfermedad se induce mediante la administración a la rata de Acs anti-MBG producidos en otra especie, a la que se ha inmunizado previamente con un preparado de MBG de rata. Este modelo fue estudiado por Masugi, y el término de nefritis nefrotóxica o nefritis de Masugi ha sido utilizado durante muchos años. Posteriormente, se demostró que la MBG contenía el Ag nefritogénico responsable de la glomerulonefritis (GN) que se desarrolla por lo que pasó a denominarse, tanto el modelo como su contrapartida en humanos, GN por Acs anti-MBG (7). La administración pasiva de Acs anti-MBG a la rata se desarrolla en dos fases: una fase aguda (heteróloga), que comienza a los pocos minutos de la administración del antisuero y una fase tardía (autóloga), que ocurre a los 7-10 días cuando la rata produce sus propios Acs y reacciona con los Acs extraños previamente depositados. El glomérulo es la principal diana de daño durante la fase heteróloga del modelo, aunque se acompaña de una respuesta inflamatoria celular intersticial de variable intensidad. El daño glomerular resulta de la activación de un número variado de mediadores de daño inmunológico. El desarrollo de proteinuria sólo ocurre tras la fijación o depósito del Ac a la MBG. Si el Ac fija complemento (C), se produce además una infiltración pasajera del glomérulo por neutrófilos a los pocos minutos. Este acúmulo de neutrófilos es máximo entre las 4-6 primeras horas, siendo posteriormente sustituido por células de estirpe linfomonocitario. Los enzimas procedentes de ambas poblaciones celulares son capaces de dañar la MBG, fragmentarla y ocasionar hematuria. No es infrecuente la formación de semilunas, siendo éstas más habituales en el conejo que en la rata. Mediante inmunofluorescencia se detecta un depósito continuo y lineal de IgG heteróloga a lo largo de la MBG y depósitos de C3 procedente del propio huésped, así como fibrina en el componente extracapilar. La fase autóloga es la consecuencia natural de la respuesta inmune del huesped al depósito de la IgG heteróloga (y por tanto ajena al mismo) en la MBG. Cantidades tan pequeñas como 5 µg de IgG con actividad anti-MBG es suficiente para inducir la fase autóloga de la enfermedad en la rata, durante la cual ésta desarrolla Acs propios contra la IgG heteróloga previamente depositada.
La forma activa se induce mediante la inmunización al animal con Ags de MBG homólogos o heterólogos en adyuvante completo de Freund. El modelo clásico fue desarrollado por Steblay utilizando como huésped la oveja, animal muy susceptible que desarrolla un GN extracapilar rapidamente progresiva, falleciendo entre 1-3 meses (8). La inmunización a ratas con preparados de MBG heteróloga da lugar al desarrollo de una GN por Acs anti-MBG de diversa intensidad, siendo ésta más severa en la cepa de ratas Brown Norway (BN) . Por otra parte, la cepa de ratas WKY a las que se inmuniza con Ags de MBG, responden con una GN extracapilar y una respuesta inmune celular acompañante (figs. 1b, c y d) (9).
Ags de las células propias del glomérulo
A. Patología en Humanos. El mecanismo exacto responsable de la formación de depósitos inmunes es desconocido en la mayoría de las GN en humanos. La existencia de Acs en el lupus murino que tienen capacidad para unirse a ciertas estructuras glomerulares, sugiere que un mecanismo similar pudiera ocurrir en humanos. Se ha demostrado la existencia de Acs anti-endotelio en casos de LED, en diversas enfermedades del tejido conectivo, y en algunas formas de vasculitis en humanos. La GN membranosa asociada a neoplasias o infecciones, semeja a la GN del modelo de la enfermedad del suero y, aun más, a la del modelo conocido como nefritis de Heymann. Ocasionalmente, se ha demostrado la presencia de Acs anti-célula mesangial en pacientes con nefropatía IgA (10,11).
B. Modelos experimentales.
Ags que se expresan en las células mesangiales:
El modelo utilizando Acs anti-Thy-1 se desarrolló una vez que se observó que Thy-1, que se expresa en los linfocitos T, estaba presente en la célula mesangial de rata (y no en otra especie). Diversos grupos de investigadores han usado indistintamente un antisuero policlonal anti-timocitos o un Ac monoclonal anti-Thy-1(OX7) (12,13). La administración del antisuero produce en la rata, tras la fijación de C, una lisis del mesangio y tras un corto período de tiempo, una reparación a través de una respuesta proliferativa de la célula mesangial. La lesión de lisis mesangial es dependiente de C, ya que su depleción evita la mesangiolisis. Además, se han observado depósitos de la fracción C5b-9 del C, el denominado complejo de ataque a la membrana (CAM), confirmando la implicación del C en el desarrollo de este modelo experimental (14). La fase de mesangiolisis ocurre entre 1 hora y 2 días, y la de reparación entre los días 3 y 5 de la enfermedad. En esta segunda fase es frecuente observar una infiltración del ovillo glomerular por linfocitos T y monocitos-macrófagos (figs. 2a y b). Finalmente, y en un tiempo que varía entre 3 y 6 semanas, se produce una resolución del proceso con esclerosis de la matriz mesangial de variable intensidad. Este modelo se ha utilizado preferentemente para estudiar la respuesta mesangial proliferativa tras el daño glomerular, así como otras diversas funciones de la célula mesangial, incluyendo sus propiedades hemodinámicas y fagocíticas (15-16).
![]()
Fig. 2. Nefritis por Ac anty-Thy-1.
A) mesangiolisis (PAS x 250) y B) fase proliferativa (HE x 450).
Ags que se expresan en el endotelio glomerular:
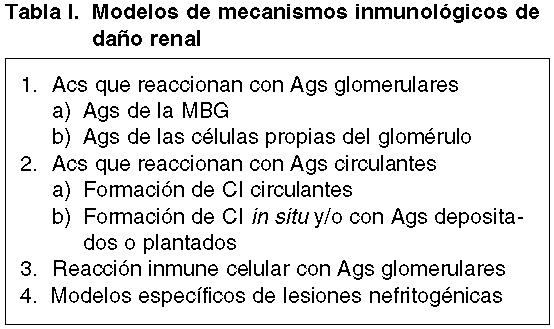
Es conocida la importancia de la interacción leucocito/célula endotelial en la extravasación de las células hematopoyéticas desde la circulación hasta el foco de lesión tisular durante la respuesta inflamatoria (17). El daño sobre la célula endotelial tiene implicaciones conocidas en la inducción de trombosis, como ocurre en el síndrome urémico hemolítico. Se han desarrollado modelos en los que se utiliza como Ag la enzima que convierte la angiotensina (ECA), que se expresa en la superficie de la célula endotelial, y administración de un Ac anti-ECA. Es sabido que la existencia de Acs anti-endotelio preformados en el huésped y su interacción con Ags que se expresan en la superficie del lecho vascular del riñón del donante, son la causa determinante del rechazo hiperagudo (fig. 3) (18).
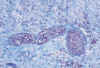
Fig. 3. Rechazo hiperagudo. Acs
anti-endotelio vascular (Masson x 480).
Ags que se expresan en la célula epitelial:
El modelo de nefritis de Heymann en la rata, es aplicable para el estudio de la GN membranosa en humanos (fig. 4a). Fue descrito por vez primera en 1959 por Heymann utilizando una forma activa de inmunización con preparados de riñon de rata en adyuvante completo de Freund (19). Posteriormente se utilizó la denominada Fx1A (fracción proteica obtenida del borde en cepillo del túbulo proximal) que produce proteinuria a las 4-6 semanas de la inmunización, fijación del Ac y C en las paredes de los capilares glomerulares, así como depósitos electrodensos en la vertiente subepitelial de la membrana basal glomerular y un patrón granular por inmunofluorescencia (figs. b,c y d) (20,21). En un principio se pensó que la nefritis de Heymann era secundaria a la formación de CI Fx1A/anti-Fx1A. Investigaciones posteriores demostraron que la lesión era producida por la unión del Ac circulante al Ag Fx1A presente en las uniones de los pies de los podocitos. Durante muchos años se ha investigado sobre la exacta naturaleza del Ag/s nefritogénico de Heymann presente en la Fx1A. Estos estudios han demostrado que el principal Ag es una glicoproteína de 330 kD (gp330), recientemente denominada megalina (22), y que se asemeja al receptor de las lipoproteínas de baja densidad (LDL), siendo capaz de formar complejos con otra proteína más pequeña denominada proteína asociada al receptor (RAP) (23,24). La unión del Ac a la membrana celular del podocito es seguida de la activación del C con separación, recubrimiento y diseminación de los agregados Ag/Ac por la superficie de la célula epitelial, formando los característicos depósitos subepiteliales.

Fig. 4. A) Nefropatía Membranosa.
Depósitos granulares subepiteliales de IgG (IF x 600); B) Nefritis de Heymann
(IF x 350); C) Ag de Heymann (borde en cepillo del epitelio tubular), (IF x 250)
y, D) Depósitos electro-densos subepiteliales (ME x 4.580).
Acs QUE REACCIONAN CON Ags CIRCULANTES
A. Patologia en Humanos. La mayoría de las GN en humanos muestran CI en el glomérulo, generalmente localizados en determinadas estructuras (mesangio, pared capilar), conteniendo clases específicas de Ig. Aunque los modelos experimentales caracterizados por depósitos de Ig, se sabe que interaccionan con Ags estructurales, se asume que la mayoría de ellos representan CI depositados o formados in situ (Ags plantados) a los que posteriormente se une el Ac. Entre este tipo de GN mediada por CI se incluyen: GN proliferativa mesangial, GN membranosa, GN membranoproliferativa y GN proliferativa extracapilar. En otras nefropatías en humanos, la enfermedad se define por el tipo de depósito inmune como: la nefropatía IgA y la nefropatía IgM. Aunque en la mayoría de los casos no se identifican los Ags solubles responsables de la enfermedad glomerular, en los casos asociados a infecciones, neoplasias, medicamentos o enfermedades autoinmunes, se han identificado los Ags en los depósitos glomerulares (25,26).
B. Modelos Experimentales.
Formación de CI circulantes:
La enfermedad aguda del suero inducida tras la administración al conejo de una única dosis de la proteína ajena BSA (albúmina sérica bovina), es el mejor ejemplo de enfermedad por CI circulantes (revisado en 27,28). El animal desarrolla Acs anti-BSA a los 4-5 días de la administración de BSA y forma complejos BSA/anti-BSA estando el Ag en exceso en la circulación. Al ser los CI circulantes de pequeño tamaño, éstos no se agregan y no pueden ser fácilmente eliminados por el sistema mononuclear-fagocítico. Conforme aumenta la respuesta inmune y existe una mayor cantidad de Acs anti-BSA en la circulación, la relación Ag/Ac se invierte y aumenta el tamaño de los CI, que son más fácilmente fagocitados y eliminados. Durante este proceso, una pequeña fracción de los complejos BSA/anti-BSA se acumula en los glomérulos y otros lechos vasculares, producciendo una GN aguda y lesiones de vasculitis. La severidad del daño glomerular va a estar en relación con el tamaño de los CI, la avided del Ac, liberación de substancias vasoactivas, aclaramiento de los CI por el sistema mononuclear-fagocítico, y otros parámetros que contribuyen a favorecer su depósito en el glomérulo. Este acúmulo de CI en el glomérulo se acompaña de un infiltrado de neutrófilos y de células mononucleadas, así como de proteinuria. Los estudios por inmunofluorescencia muestran al inicio un patrón finamente granular de depósitos de Ig, C y BSA en las paredes capilares del glomérulo renal. Más tarde, y coincidiendo con un exceso de Acs en la circulación (al aumentar la respuesta inmunológica del animal), se hacen más intensos los depósitos de Ig mientras disminuyen los de BSA, para posteriormente acabar de desaparecer la proteinuria (fig. 5 a y b).
![]()
Fig. 5. Enfermedad aguda del suero:
A) Depósitos granulares subepiteliales de IgG (IF x 550) y B) Depósitos
electrodensos de localización subepitelial (ME x 5.600).
La enfermedad crónica del suero se induce mediante la inyección diaria y durante varias semanas de proteínas ajenas al animal inmunizado. Se produce de esta forma una respuesta inmunológica con formación de Acs y complejos Ag/Ac circulantes, así como depósitos glomerulares conteniendo la proteína administrada y los Acs producidos por el huésped. Dependiendo de la capacidad de respuesta inmune del animal inmunizado y de la cantidad de Ag administrado, las lesiones histológicas de la enfermedad crónica del suero inducida en el conejo por la administración de BSA, son muy variables y se asemejan a algunas formas de GN en humanos. Los animales que responden con una alta producción de Acs anti-BSA desarrollan una GN extracapilar rápidamente progresiva, mientras que aquéllos otros que tienen una discreta respuesta inmunológica muestran diversas formas histológicas de GN proliferativa con infiltrados celulares inflamatorios en el glomérulo, engrosamiento de la pared capilar, así como una proteinuria severa. Aunque la localización del los CI depositados, conteniendo el Ag, Ac y C, es preferentemente subepitelial, también se pueden localizan en la propia membrana basal y en el mesangio (29,30). En aquellos animales que tienen una respuesta inmune muy alta, y que por tanto producen una gran cantidad de CI circulantes, éstos también se acumulan en el lecho vascular sistémico de una forma muy similar al que ocurre en el LED en humanos (fig. 6 a, b y c) (31).
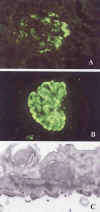
Fig. 6. Enfermedad crónica del
suero: A) Patrón mesangial (IF x 350); B) Patrón subepitelial (IF x 350) y, C)
Depósitos subepitaliales (ME x 8.600).
Formación de CI in situ y/o con Ags depositados o plantados:
Un hecho importante relacionado con los mecanismos de formación de los CI, es la interacción in situ de los componentes proteicos que forman los depósitos glomerulares con los otros componentes también proteicos (ya libres o formando complejos) en la circulación (32). Cuando el otro miembro se une a su par correspondiente (Ag/Ac versus Ac/Ag) , se forma un nuevo acúmulo de material inmune in situ sobre los CI previamente depositados, ampliando de esta forma el daño glomerular. Otro mecanismo responsable de la formación in situ de CI ocurre cuando un Ac se une a un Ag localmente en el glomérulo (33). Los Acs circulantes pueden unirse con Ags estructurales del glomérulo pero también con Ags que se han depositado previamente. Esta segunda posibilidad se ha demostrado en estudios utilizando la concanavalina A (ConA). Al ser una lectina, ésta se une a glicoproteínas ricas en manosa depositándose en el glomérulo. Una vez «plantada», la ConA actúa como un Ag capaz de unirse a un Ac. Otras proteínas (albúmina e IgG cationizadas), varias lectinas, moléculas policatiónicas como heparina, laminina, proteoglicanos, integrinas de la superficie del epitelio visceral y ECAS en el endotelio, etc., se han utilizado para el estudio de la formación in situ de CI en el glomérulo renal (34).
El extenso número de modelos experimentales desarrollados hasta la fecha con el fin de estudiar los mecanismos responsanbles de la formación de CI (tanto circulantes como in situ) así como la amplia variedad de Ags utilizados, hace que su completa descripción sobrepase la finalidad para la que fue orientado el presente trabajo. No obstante, y con el fin de completar este estudio, se comentarán los mecanismos de formación de CI en el modelo murino de LED. Fue en la cepa de ratones NZB donde primero se observó el desarrollo de una GN lúpica que aparecía en edades avanzadas, falleciendo los animales entre los 18-24 meses de vida. El cruce resultante de esta cepa con la NZW, NZBxNZW F1, conlleva un comienzo más temprano de la GN y al fallecimiento entre los 8-12 meses, siendo en ambas cepas la enfermedad más grave en las hembras (35). Posteriormente, se observó que los ratones MRL/lpr presentaban una mayor hiperplasia linfoide y un comienzo más temprano de la GN con fallecimiento a los 8 meses (36,37). Existen numerosos estudios que indican que hay diversos mecanismos implicados en la formación de CI en el LED (38). Se sabe que el ADN tiene capacidad para unirse a la pared capilar del glomérulo renal y servir como lugar para la formación de CI in situ. El ADN o los complejos ADN/anti-ADN también reaccionan con la fibronectina. Más recientenmente, se ha postulado el papel del complejo histona-ADN (nucleosomas) y su Ac, como los CI que se depositan en el glomérulo mediante un mecanismo dependiente de carga y unión a heparán sulfato, un componente de la MBG (39). Un hecho que apoyaría el papel de las histonas en esta enfermedad, es la presencia de estas proteínas en los depósitos glomerulares de ratones NZBxNZW F1. Además de los mecanismos implicados en la formación de CI circulantes e in situ en el LED, se ha visto que los Acs obtenidos de eluidos de ratones con GN lúpica se unen directamente mediante reacción cruzada a los componentes estructurales de la MBG (figs. 7a y b) (40).
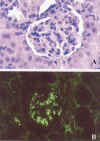
Fig. 7. Lupus murino MR/lpr: A)
Proliferación mesangial (HE x 480) y B) Depósitos mesangiales de IgG (IF x
350).
REACCIÓN INMUNE CELULAR CON AGS GLOMERULARES
A. Patología en Humanos. La mayoría de las enfermedades glomerulares en humanos se caracterizan por la presencia de depósitos de Ig de diversas clases. Sin embargo, la existencia de una respuesta celular estaría apoyada en base a la frecuente presencia de linfocitos T y monocitos-macrófagos en las lesiones glomerulares. No existe duda de que los dos brazos de la respuesta inmune —humoral y celular— están implicados en el inicio de la respuesta inmune nefritogénica. También se ha postulado que alteraciones de las células T podrían jugar un papel en el desarrollo de la enfermedad de cambios mínimos y/o esclerosis focal y segmentaria, frecuente en niños (41).
B. Modelos Experimentales. En una gran mayoría de modelos es suficiente una respuesta humoral para producir daño glomerular. Un ejemplo de ello es la rápida respuesta inflamatoria que ocurre a los pocos minutos de la administración pasiva de Acs-anti-MBG a la rata. No obstante, e incluso en esta situación, los linfocitos T al inicio y más tarde células de estirpe monocitario, forman parte de la respuesta inmunológica (42). En la cepa de ratas WKY existen datos que sugieren la existencia de mecanismos efectores específicos de una respuesta inmune celular. La transferencia de la enfermedad mediante linfocitos en ausencia de Acs, apoya el papel de la inmunidad celular como mecanismo nefritogénico (43). Un modelo interesante de GN producida por un mecanismo inmune celular es el desarrollado en aves inmunizadas con MBG heteróloga. En estas aves, es posible transferir la lesión glomerular a otro animal singénico mediante linfocitos sensibilizados (44). La administración pasiva de un antisuero heterólogo anti-MBG o la inmunización activa a ratas WKY, produce una GN extracapilar que se acompaña de un infiltrado mononuclear importante, así como de un aumento de la actividad NK. La depleción de la subpoblación de linfocitos T CD8+, así como el tratamiento con Acs anti-CD5 o anti-CD4, modulan la formación del componente extracapilar glomerular (45).
Finalmente, se ha sugerido una relación entre la inmunidad celular y la enfermedad de cambios mínimos en humanos. Factores procedentes del suero o de linfocitos T CD4+ de pacientes con esta enfermedad producen un incremento de la permeabilidad capilar y alteraciones en los pedicelos de los podocitos glomerulares y en el revestimiento aniónico de la pared capilar, tras su administración por la arteria renal (46).
MODELOS ESPECÍFICOS DE LESIONES GLOMERULARES NEFRITOGÉNICAS
Nefropatía IgA
A. Patología en Humanos. La nefropatía IgA, es una de las formas más comunes de GN en humanos, caracterizada por la presencia de depósitos de IgA, C3, y a menudo de IgG en el mesangio. Se asocia con un variable grado de proliferación mesangial y antecedentes clínicos de hematuria macroscópica coincidiendo con un cuadro infeccioso. La etiología no es conocida completamente, habiéndose postulado como posible causa, ciertas abnormalidades en el control de la producción de la IgA, preferentemente polimérica. El acúmulo de depósitos de IgA podría representar CI cuyos Ags tuvieran un origen alimentario, infeccioso, o formar parte de las propias estructuras glomerulares (47,48).
B. Modelos Experimentales. Son numerosos los modelos experimentales desarrollados en relación con la formación de CI en este tipo de nefropatía. Uno de los modelos inicialmente utilizado fue la administración, tanto oral como sistémica de dextrano. Otros investigadores han utilizado la administración de agregados de IgG o IgA. La cepa de ratones ddY desarrollan espontáneamente con la edad una nefropatía con depósitos de IgA. Los depósitos glomerulares coinciden con una elevación de la IgA sérica. La infección por el virus Sendai induce un modelo de nefropatía IgA muy parecida a la que se desarrolla en humanos tras un cuadro infeccioso (revisado en 49). Finalmente, se observó que los depósitos de IgA en el mesangio se asocian a diversas formas de hepatopatía. Así, los depósitos de IgA se pueden ver en el daño hepático asociado al tetracloruro de carbono o tras la ligadura del conducto biliar. El alcohol es otro agente capaz de producir depósitos del componente secretorio de la IgA. Estos hallazgos son muy parecidos a lo que ocurre en pacientes con cirrosis alcohólica o atresia biliar, en los que el componente secretorio de la IgA se detecta en el mesangio (50).
GN Necrotizante Extracapilar «pauci-inmune»
A. Patología en Humanos. La denominada GN necrotizante extracapilar, que se asocia a la granulomatosis de Wegener y a otras formas de vasculitis, representa un tipo de GN severa con ausencia o una muy pequeña cantidad de depósitos de CI. Frecuentemente se detectan Acs anti-Ags del citoplasma de los neutrófilos (ANCAS), describiéndose dos patrones: a) citoplásmico, denominado c-ANCA, en el que el Ag reactivo es la proteinasa-3 y b) perinuclear o p-ANCA, que reacciona con la mieloperoxidasa (MPO). c-ANCA se asocia con la granulomatosis de Wegener y p-ANCA con otros tipos de vasculitis, incluyendo aquéllas con Acs anti-MBG (revisado en 51).
B. Modelos Experimentales. La administración a ratas de MPO humana (que presenta reacción cruzada con la de rata), induce una respuesta inmune humoral y celular. La perfusión a través de la arteria renal con MPO a ratas preinmunizadas con extractos de neutrófilos y peróxido de hidrógeno, produce depósitos de MPO e Ig en la MBG y una respuesta inflamatoria granulomatosa de los vasos de pequeño calibre (52). En la enfermedad aguda del suero y en las cepas de ratones que desarollan espontáneamente LED, es frecuente el hallazgo de lesiones de vasculitis necrotizante (53). Anti-MPO-ANCAS es un hallazgo habitual en el modelo de nefritis autoinmune en la rata Brown Norway inducida por la administración de mercurio (fig. 8) (54).
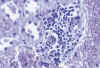
Fig. 8. Nefritis intersticial
autoinmune.
Síndrome urémico hemolítico/púrpura trombótica trombocitopénica
A. Patología en Humanos. El síndrome urémico hemolítico (SUH) y la púrpura trombótica trombocitopénica (PTT) son formas de presentación clínica asociadas con situaciones que producen daño en el endotelio vascular y consecuentemente, microangiopatía trombótica (MAPT). El cuadro clinicopatológico incluye: anemia hemolítica, tombocitopenia, fracaso renal, y microtrombosis de las arteriolas y capilares glomerulares del parénquima renal. La afectación del sistema nervioso central es un hecho más en relación con la PTT. El SUH/PTT se asocia con diarrea, tóxicos, embarazo y neoplasias, así como con formas familiares. Recientemente se ha demostrado el papel determinante de ciertas toxinas producidas por algunas cepas de E.coli, como la denominada «Shiga-like» (SLT) y la verotoxina (VT) (55).
B. Modelos Experimentales. Los intentos de producir lesiones de MAPT en los glomerulos utilizando VT, no han sido muy definitivos hasta la fecha. La administración de VT a primates produce un cuadro parecido al SUH. En este modelo, se produce una elevación de la endotelina sin un incremento compensatorio de la producción de óxido nítrico, y lesiones, en un pequeño porcentaje de animales de MAPT glomerular (56).
BIBLIOGRAFÍA
Wilson CB, Cole E, Zanetti M, Mampaso F. Renal Diseases. In «Basic and Clinical Immunology». Edited by Lange, Los Altos, California, 1982; 557-75.
Hoedemaeker PJ, Ten J, Hogeendorn PC, Kawasaki K, van Leer EH, de Heer E, Fleuren GJ. Pathogenesis of glomerulonephritis: experimental models revisited. Adv Nephrol NeckHosp 1991; 20: 73-90.
Wilson CB. Renal response to immunologic glomerular injury. In: Brenner BM, ed. The Kidney, 5th ed. Philadelphia: Saunders, 1996; 1253-391.
Couser WG. New insights into mechanisms of immune glomerularinjury. West J Med 1994; 160: 440-6.
Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med 1967; 126: 989-1004.
Kalluri R, Wilson CB, Weber M, Gunwar S, Chonko AM, Neilso EG, Hudson BG. Identification of the a3 chain of type IV collagen as the common autoantigen in anti-basement membrane disease and Goodpasture syndrome. J Am Soc Nephrol 1995; 6: 1178-85.
Dixon FJ, Wilson CB. The development of immunopathologic investigation of kidney disease. Am J Kidney Dis 1990; 16: 574-8.
Steblay RW. Glomerulonephritis induced in sheep by injections of heterologous glomerular basemen membrane and Freund’s complete adjuvant. J Exp Med 1962; 116: 253-72.
Pusey CD, Holland MJ, Cashman SJ, Simico RA, Lloveras JJ, Evans DJ, Lockwood CM. Experimental autoimmune glomerulonephritis induced by homologous and isologous glomerular basement membrane in Brown Norway rat. Nephrol Dial Transplant 1991; 6: 457-67.
Cines DB, Lyss AP, Reeber M, Bina M, DeHoratius RJ. Presence of complement-fixing anti-endothelial cell antibodies in systemic lupus erythematous. J Clin Invest 1984; 73: 611-25.
D’Cruz DP, Houssiau FA, Ramirez G, Baguley E, McCucheon J, Vianna J, Haga H, Swana GT, Hughes GRW. Antibodies to endothelial cells in systemic lupus erythematous: A potential marker for nephritis and vasculitis. Clin Exp Immunol 1991; 85: 254-61.
Yamamoto T, Wuilson CB. Quantitative and qualitative studies of antibody-induced mesangial cell damagein the rat. Kidney Int 1987; 32: 514-25.
Bagchus WM, Hoedemaeker PJ, Rozing J, Bakker WW. Acute glomerulonephritis after intravenousinjection of monoclonal anti-thymocyte antibodies in the rat. Immunol Lett 1986; 12: 109-13.
YamamotoT, Wilson CB. Complement dependence of antibody-induced mesangial cell injury in the rat. J immunol 1987; 138: 3758-65.
Johnson RJ, Iida H, Alpers CE, Majesky MW, Schwarz SM, Prizl P, Gordon K, Gown AM. Expression of smoth muscle phenotype by rat mesangial cells in immune complex nephritis a-smooth muscle actin is a marker of mesangial cell proliferation. J Clin Invest 1991; 87: 847-58.
Yamamoto T, Mundy K, Wilson CB, Blantz RC. Effect of mesangial cell lysis and proliferation on glomerular hemodynamics in the rat. Kidney Int 1991; 40: 705-13.
Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol 1993; 11: 767-804.
Pall AA, Savage CO. Mechanisms of endothelial cell injury in vasculitis. Springer Semin Immunopathol 1994; 16: 23-37.
Heymann W, Hackel DB, Harwood S, Wilson SGF, Hunter JLP,. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc Soc Esp Biol Med 1959; 100: 660-4.
Edgington TS, Glassock RJ, Dixon FJ. Autologous immune complex nephritis induced with renal tubular antigen. I. Identification and isolation of the pathogenic antigen. J Exp Med 1968; 127: 555-71.
Glassock RJ, Edgington TS, Watson JI, Dixon FJ. Autologous immune complex nephritis induced with renal tubular antigen. II. The pathogenic mechanisms. J Exp Med 1968; 127: 573-87.
Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol 1995; 6: 35-47.
Raychowdhury R, Niles J, McClusky RT, Smith JA. Autoimmune target in Heymann nephritis is a glycoporotein with homology to the LDL-receptor. Science 1989; 244: 1163-5.
Strickland DK, Ashcom JD, Williams S, Battey F, Behre E, McTigue K, Battey JF, Argraves WS. Primary structure of (2-macroglobulin receptor-associated protein. J Biol Chem 1991; 266: 13364-9.
Kashgarian M. Lupus nephritis: lessons for the path lab. Kidney Int 1994; 45: 928-38.
Wilson CB. The renal response to immunologic injury. In: Brenner BM, Rector FC . eds. The Kidney, 4th ed. Philadelphia: Saunders, 1991; 1: 1062-181.
Germuth FG Jr, Rodriguez E, Lorelle K, Trump EI, Milano L, Wise O. Passive immune complex glomerulonephritis in mice: Models for various lesions found in human disease. I. High avidity complexes and mesangipathic glomerulonephritis. Lab Invest 1979; 41: 360-5.
Germuth FG Jr, Rodriguez E, Lorelle K, Trump EI, Milano L, Wise O. Passive immune complex glomerulonephritis in mice: Models for various lesions found in human disease. II. Low avidity complexes and diffuse proliferative glomerulonephritis with subepithelial deposits. Lab Invest 1979; 41: 366-71.
Germuth FG Jr, Rodriguez E. Immunopathologic of the Renal Glomerulus: Immune Complex Deposit and Antibasement Membrane Disease. Boston: Little Brown, 1973.
Dixon FJ, Feldman JD, Vazquez JJ. Experimental glomerulonephritis. The pathogenesis of a laboratory model resembling the sprectum of human glomerulonephritis. J Exp Med 1961; 113: 899-920.
Neuland C, Albini B, Brentjens J, Groosberg AI, Andres GA. Antigen concentration in tissues of rabbits with systhemic chronic serum sickness. Int Arch Allergy Appl Immunol 1981; 64: 385-394.
Ward DM, Lee SS, Wilson CB. Direct antigen binding to glomerular immune complex deposits. Kidney Int; 1986; 30: 706-11.
Ward HJ, Cohen AH, Border WA. In situ formation of subepithelial immune complexes in the rabbit glomerulus: Requierement of a cationic antigen. Nephron 1984; 36: 257-64.
Mannik M, Gauthier VJ, Stapleton SA, Agodoa LYC. Immune complexes with cationic antibodies deposit in glomeruli more effectively than cationic antibodies alone. J Immunol 1987; 38: 4209-17.
Howie JB, Helyer BJ. The immunology and pathology of NZB mice. Adv Immunol 1968; 9: 215-66.
Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice caused by a point mutation in the Fas ligand. Cell 1994; 76: 969-76.
Pérez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, Schlondorff D. Chemokine expression precedes inflammatory cell infiltration, chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol 2001; 12: 1369-82.
Theofilopoulos AN, Dixon FJ. Murine models of systhemic lupus erythematous. Adv Immunol 1985; 37: 269-358.
Schmiedeke TMJ, Stockl FW, Weber R, Sugisaki Y, Batsford SR, Vogt A. Histones have high affinity for the glomerular basement membrane. J Exp Med 1989; 169: 1879-94.
Vogt A, Batsford SR, Morioka T. Nephritogenic antibodies in lupus nephritis. Tohoku J Exp Med 1994; 173: 31-41.
Atkins RC, Holdsworth SR. Cellular mechanisms of immune glomerular injury. In: Wilson CB, Brenner BM, Stein J,eds. Contemporary Issues in Nephrology, vol 18, New York: Churchill Livingston, 1988; 111-35.
Kreisberg JI, Wayne DB, Karnovsky MJ. Rapid and focal lossof negative charge associated with mononuclear cell infiltration early in nephrotoxic serum sickness. Kidney Int; 1979; 16: 290-300.
Tipping PG, Neale TJ, Holdsworth SR. T lymphocyte participation in antibody-induced experimental glomerulonephritis. Kidney Int 1985; 27: 530-7.
Bolton WK, Tucker FL, Sturgill BC. Experimental autoimmune glomerulonephritis in chickens. J Clin Lab Immunol 1980; 3: 179-84.
Kawasaki K, Yaoita E, Yamamoto T, Kihara I. Depletion of CD8 positive cells in nephrotoxic serum nephritis of WKY rats. Kidney Int 1992; 41: 1517-26.
Wilkinson AH, Gillespie C, Hartlty B, Williams DG. Increase in proteinuria and reduction in number of anionic sites on the glomerular basement membrane in rabbits by infusion of human nephrotic plasma in vivo. Clin Sci 1989; 77: 43-8.
Emanmcipator SN. Immunoregulatory factors in the pathogenesis of IgA nephropathy. Kidney Int 1990; 38: 1216-29.
van Es LA. Pathogenesis of IgA nephropathy. Kidney Int 1992; 41: 1720-9.
Rifai A. Experimental models for IgA-associated nephritis. Kidney Int 1987; 31: 1-7.
Smith SM, Leaber R, Lefebre A, Leung MF, Baricos WH, Leung WC. Pathogenesis of IgA nephropathy in ethanol comsumption:animal model and cell culture studies. Alcohol 1993; 10: 477-80.
Jennette JC, Falk RJ. Antineutrophil cytoplasmic autoantibodies and associated diseases: a review. Am J Kidney Dis 1990; 15: 517-29.
Brouwer E, Huitema MG, Klok PA de Weerd H, Cohen T, Weening JJ, Kallemberg CGM. Antimyeloparoxidase-associated proliferative glomerulonephritis: an animal model. J Exp Med 1993; 177: 905-14.
Kinjoh K, Kayogoku M, Good RA. Genetic selection for crescent formation yields mouse strai with rapidly progressive glomerulonephritis and small vessel vasculitis. Proc Natl Acad Sci USA 1993; 90: 3413-7.
Kobayashi K, Shibata T, Sugisaki T. Aggravation of rat Masugi nephritis by heterologous anti-rat myeloparoxidase (MPO) antibody. Clin Exp Immunol 1993; 93 (Suppl 1): 20.
Richardson SE, Karmali MA, Becker LE, Smith CR. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections . Human Path 1988; 19: 1102-8.
Siegler RL, Taylor FB, Tesh VL, Edwin SS, Cook JB, Dudley DJ. The endothelin-nitric oxide axis in a primate model of shiga-like toxin induced hemolytic uremic syndrome. J Am Soc Nephrol 1995; 6: 989-97.
![]()