
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
Antonio Barat, Mar del Barrio Molina, Félix Manzarbeitia
Dpto. de Anatomía Patológica. Fundación Jiménez Díaz UAM. Madrid*
* Este capítulo lo dedicamos a la memoria de la Srta. Marilis Lagunar, técnico de microscopia electrónica de nuestro departamento (1991†).
I. INTRODUCCIÓN
A pesar de que el desarrollo de las técnicas inmunohistoquímicas ha provocado un notable descenso en la aplicación de la microscopía electrónica, ésta sigue siendo fundamental en el estudio de la patología renal. Junto con los métodos inmunopatológicos, constituye el complemento fundamental de la microscopía óptica para el diagnóstico de toda biopsia renal, al menos en cuanto a la enfermedad glomerular. Hay que señalar que la patología renal fue una de las aplicaciones iniciales de la microscopía electrónica en medicina, contribuyendo de forma fundamental a su diagnóstico y clasificación. En nuestro país estos estudios se iniciaron por el Dr. Horacio Oliva, que implantó la técnica en nuestro departamento, aplicándola fundamentalmente a la patología renal, histogénesis de tumores y procesos linfoproliferativos. Parte de esta experiencia inicial, con el Dr. Vicente Navarro, se tradujo en la publicación de un libro (1). Posteriormente, junto con los conocimientos de inmunofluorescencia en patología renal de un autor (AB) (2), dio lugar a la publicación de otro libro (3). Parte de las imágenes que ilustran este capítulo proceden de la experiencia entonces acumulada (4-7).
II. ASPECTOS METODOLÓGICOS
Para una correcta valoración de la patología renal (8-14), hay que realizar un procesamiento muy riguroso de las biopsias renales, ya que una manipulación incorrecta conlleva la destrucción de la información que podría aportar el material biópsico e incluso dar lugar a la emisión de un diagnóstico erróneo. Por lo general, en la actualidad, las biopsias corresponden a finos cilindros, ya que se obtienen mediante punción con aguja fina. El tejido del que se puede disponer es muy escaso, y debe manejarse de forma adecuada para obtener el máximo rendimiento. Para ello, es esencial conocer la información clínica, lo que permitirá priorizar la división del tejido en fragmentos destinados a los diferentes estudios. La visualización de glomérulos mediante la observación con el microscopio estereoscópico permitirá una mejor selección de las porciones tisulares. Es obligada la toma de muestras para microscopía de inmunofluorescencia, y un pequeño fragmento de 1 mm. para microscopia electrónica. En las biopsias obtenidas por lumbotomía por lo general no hay problema en la toma de muestras para los diferentes métodos de estudio.
Las biopsias se remitirán lo más rápidamente posible en una gasa empapada en suero fisiológico y a ser posible, deben obtenerse dos cilindros. En caso de no haber suficiente material y la clave diagnóstica pueda encontrarse en el estudio ultraestructural, se procederá a coger muestras para inmunofluorescencia y microscopía electrónica. Posteriormente, se puede intentar recuperar el material sobrante de la inmunofluorescencia destinándolo para microscopía óptica, mediante fijación en formol. El proceder de reutilizar el material de inmunofluorescencia para microscopía óptica da buenos resultados si la recuperación del material se hace antes de las 48 horas después de la congelación para inmunofluorescencia.
En algunos casos, por el hallazgo de lesiones que necesitan comprobación ultraestructural, se puede destinar el material incluido en parafina o el sobrante de la inmunofluorescencia para microscopía electrónica. Aunque se producen importantes artefactos, el tejido puede estar lo suficientemente conservado como para permitir la visualización de muchas de las alteraciones ultraestucturales (como ejemplo, ver fig. 14).
La muestra destinada para microscopía electrónica debe ser fijada en glutaraldehido con postfijación en tetróxido de osmio, e inclusión posterior en resina sintética. Antes de la observación con el microscopio electrónico se deben hacer cortes semifinos de control para tener una visión panorámica y poder escoger las áreas apropiadas para el estudio ultraestructural. Éste debe ser sistemático e incluir todos los compartimientos: glomérulos, túbulos, vasos e intersticio, si bien en la mayoría de los casos es imprescindible la porción glomerular.
La más importante aportación de la microscopía electrónica se encuentra en las enfermedades glomerulares mediadas por complejos inmunes, ya que permite localizar con exactitud los depósitos densos electrónicamente, que se corresponden con los inmunocomplejos apreciados por inmunofluorescencia y los depósitos fuchinófilos objetivados en microscopía óptica con la técnica tricrómica de Masson. Éstos, con frecuencia no se aprecian de forma clara con la microscopía óptica, por su escasa cuantía, y el estudio con inmunofluorescencia en ocasiones es dudoso, lo que convierte a la microscopía electrónica en instrumento fundamental para el diagnóstico. Es el momento de precisar que no todo depósito denso electrónicamente hay que catalogarlo como «depósito inmunológico», y no olvidar la posibilidad de que haya proteínas atrapadas en el filtro glomerular, como ocurre en la hialinosis focal.
También tiene su importante contribución la microscopía electrónica en los procesos que cursan con alteraciones de la membrana basal, como son: adelgazamiento con o sin ruptura de la misma, laminación difusa y rarefacción, muchas de ellas con rasgos genéticos (nefropatía de Alport, hematuria recidivante familiar, síndrome nefrótico congénito). Otras aportaciones son las alteraciones en la vertiente subendotelial de la membrana basal, por ejemplo en los procesos microangiopáticos y en la glomerulopatía crónica post-trasplante. También la microscopía electrónica puede permitir la visualización de partículas virales.
Haremos referencia fundamentalmente a la patología glomerular, dado que las alteraciones en los restantes compartimientos, salvo algunas excepciones, se pueden caracterizar bien por microscopía óptica.
Los pasos a seguir para la observación en patología ultraestructural son semejantes al de toda biopsia renal:
Siempre cumpliendo la premisa del conocimiento de la realidad clínica, es recomendable, primeramente, la observación del cilindro de la microscopía óptica y de la inmunofluorescencia, para tener una idea general del proceso patológico.
Estudio del corte semifino (0·5 µm) de control, teñido con Giemsa, y en algunos casos con plata Metenamina, para guiar el estudio en la fase siguiente.
Visión general de los múltiples cortes ultrafinos (microtomo LKB) montados en la rejilla, e incluso de otros bloques, dependerá del material disponible.
Dentro del glomérulo observación de:
Área mesangial y células mesangiales.
Endotelio.
Membrana basal (fig. 1), con espesor medio de 300 nm.
Célula epitelial visceral (podocitos).
Célula epitelial parietal.
Espacio de Bowman.
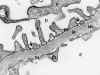
Fig. 1. Capilar glomerular: En
endotelio, Mb membrana basa (ld lámina densa, Ire lámina rara externa, lri
lámina rara interna) P Podocito, p pedicelos, flechas diafragmas de las
hendiduras de filtración, U espacio urinario.
III. SINOPSIS DE LA PATOLOGÍA ULTRAESTRUCTURAL DEL GLOMÉRULO RENAL
Es importante tener la idea clara de que los hallazgos morfológicos, salvo raras excepciones, no son específicos, ya que una misma imagen morfológica puede ser debida a varias causas; los hallazgos deben ser valorados, como cualquier tipo de biopsia o citología, en el contexto clínico. No podemos dejar de enfatizar la necesidad de disponer de la información clínica lo más completa posible.
A continuación exponemos una relación de las lesiones más habituales, sin seguir ninguna clasificación etiopatogénica ni clinicopatológica, con la intención de que sirva de referencia general. Necesariamente dada la extensión de este capítulo, para detalles más precisos el lector debe utilizar la bibliografía especializada. También debe tener en cuenta que algunos aspectos (enfermedades microangiopáticas, enfermedad de cadenas ligeras y fibrilares,...) se tratan en otros capítulos de esta monografía.
1. Cambios mínimos («Enfermedad de cambios mínimos». «Nefrosis lipoidea»)
Los «cambios mínimos» es la causa más importante del síndrome nefrótico infantil sensible al tratamiento con corticoesteroides (15). Desde el punto de vista de la microscopía electrónica las alteraciones más relevantes se localizan a nivel de los podocitos (fig. 2), y consisten en la desaparición de los pedicelos, que se manifiesta por la aposición directa del citoplasma podocitario sobre la membrana basal glomerular (fig. 3), con disminución del compartimiento entre el podocito y la lámina rara externa de la membrana basal. Se aprecian en la superficie del citoplasma del podocito, en relación con el espacio urinario, signos de transformación vellositaria. En el resto del citoplasma pueden verse vacuolas o inclusiones proteicas. Membrana basal de grosor normal con ausencia de alteraciones significativas a nivel de la lámina rara externa, lámina rara interna y lámina densa. Ausencia de depósitos electrón densos. Alteraciones del tipo de cambios mínimos con síndrome nefrótico, se pueden encontrar, entre otras enfermedades, en el linfoma de Hodgkin (fig. 4) (16,17).
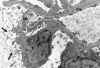
Fig. 2. Síndrome nefrótico infaltil,
cambios mínimos. Panorámica en la que se observa desaparición de los
pedicelos y transformación vellositaria de los podocitos (flechas).

Fig. 3. Síndrome nefrótico infantil,
cambios mínimos. Detalle de la desaparición de los pedicelos, con aposición
directa del citoplasma sobre la membrana basal.
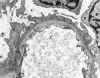
Fig. 4. Síndrome nefrótico, cambios
mínimos en pacientes con linfoma de Hodkgin. Capilar glomerular en el que se
observa desaparición de los pedicelos y transformación villositaria de los
podocitos.
La nefropatía IgM es una entidad muy controvertida, de causa desconocida, con respuesta a la terapia heterogénea y con implicaciones pronósticas poco definidas; se caracteriza por la presencia de IgM y C’3 a nivel mesangial. Al microscopio electrónico se pueden observar en algunos casos depósitos electrón densos a nivel mesangial (fig. 5) (9).
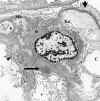
Fig. 5. Glomerulonefritis mesangial
IgM. Depósito electrón denso paramesangial (flecha).
2. Glomerulosclerosis focal segmentaria
Término que caracteriza a una lesión de distribución irregular en los glomérulos, con esclerosis de algunos de ellos (lesión focal) y de una parte del ovillo (lesión segmentaria); su aplicación debe ser utilizada con cautela, por ser un problema de difícil diagnóstico, y realizarse siempre bajo correlación clínica-patológica. Básicamente hay dos formas de presentación; a) como lesión primaria o idiopática y b) como lesión secundaria o asociada a múltiples enfermedades.
a. Glomerulosclerosis focal y segmentaria idiopática
Es la causa de lesión primaria en un 7 y 10% en niños y adultos respectivamente con síndrome nefrótico. Hay una serie de datos que la distinguen de la lesión de cambios mínimos, siendo uno de los más llamativos la escasa respuesta al tratamiento con corticoesteroides, la evolución frecuente hacia una glomerulonefritis crónica y la presencia de IgM y C’3 en los glomérulos esclerosados, que constituyen las lesiones de hialinosis desde el punto de vista óptico. Sin embargo, para algunos autores, esta lesión representa una fase evolutiva de la enfermedad por cambios mínimos y la presencia de hialinosis sería la consecuencia del atrapamiento de proteínas plasmáticas. Ultraestructuralmente se evidencia en las lesiones de hialinosis depósitos electrón densos de localización subendotelial (fig. 6), así como las lesiones del tipo de cambios mínimos como pérdida de los pedicelos de los podocitos, separación de la membrana basal con cierto aspecto laminado y transformación vellositaria de los podocitos. Las lesiones de hialinosis con frecuencia no son visibles, sobre todo cuando la biopsia, en procesos iniciales no afecta a los glomérulos de la porción yuxtamedular (18) y por la toma en la muestra para microscopía electrónica de uno de los bordes del cilindro biópsico. En las zonas de esclerosis hay aumento de la matriz mesangial, material membranoide (fig. 7), y alguna célula espumosa que contiene en su citoplasma vacuolas lipídicas.
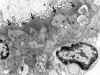
Fig. 6. Glomerulonefritis focal
segmentaria con hialinosis. Depósitos electrón densos subendoteliales en un
foco de hialinosis.
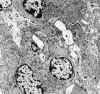
Fig. 7. Glomerulonefritis focal
segmentaria con hialinosis. Red membranoide de matriz mesangial en lesión focal
segmentaria.
b. Glomerulosclerosis focal y segmentaria secundaria
Son múltiples las causas que pueden asociarse a lesión del tipo de glomeruloesclerosis focal segmentaria (9), por lo que es un término exclusivamente morfológico que expresa sin más la existencia de una afectación glomerular ya señalada previamente, y es la correlación clínico patológica la que permite encuadrar el proceso. Para catalogar esta lesión es importante la presencia de un número suficiente de glomérulos en la biopsia, como mínimo 10. Esta lesión puede aparecer en:
Asociada a infección por virus HIV.
Nefropatía por adicción a heroína.
Obesidad.
Nefropatía por reflujo.
Agenesia renal unilateral.
Drepanocitosis.
Lupus eritematoso y vasculitis tratados.
Glomerulonefritis mesangial de IgA, como expresión de una cicatrización.
Glomerulonefritis postinfecciosa en resolución.
Nefropatía de Alport.
Síndrome nefrótico congénito.
Otras causas no especificadas.
3. Glomerulopatía membranosa («Nefropatía membranosa». «Nefropatía epimembranosa, extramenbranosa o transmembranosa»).
Causa frecuente de síndrome nefrótico del adulto (19) y de presentación poco habitual en los niños. Hay dos causas etiológicas: primaria o idiopática, de causa desconocida y secundaria asociada a diversas enfermedades como : lupus eritematoso sistémico, infecciones por virus HB, drogas (penicilamina, oro) (20) y neoplasias malignas. La lesión se caracteriza a nivel óptico por el engrosamiento difuso de las membranas basales de los capilares glomerulares, lo que se relaciona, a nivel ultraestuctural, con la presencia de depósitos electrón densos en el lado epitelial de la membrana basal, es decir entre la membrana basal y los podocitos. Se denominan comúnmente como depósitos subepiteliales o transmembranosos. Como consecuencia de estos depósitos electrón densos en la vertiente epitelial de la membrana basal e incorporación por reacción de la misma, se produce la progresión del proceso que se pueden dividir en estadios, describiéndose cuatro para la glomerulopatía membranosa según el sistema de Ehrenreich y Churg (21).
Estadio I: Membrana basal de grosor normal con ausencia o mínima presencia de púas o proyecciones membranoides de la membrana en el lado epitelial. Los depósitos subepiteliales son de pequeño tamaño (fig. 8) y la lámina densa habitualmente no muestra alteraciones; las alteraciones del citoplasma de los podocitos son poco prominentes.
Estadio II. Membrana basal gruesa con formaciones espiculares en forma de púas de peine en el lado epitelial de la misma, encontrándose los depósitos eletrón densos entre las proyecciones membranoides (fig. 9). Esta alteración de la membrana basal es muy característica, y es claramente visible con las técnicas argénticas por el carácter argirófilo de la membrana basal, y su presencia permite hacer el diagnóstico, si bién hay procesos que pueden simular una falsa imagen espicular de la membrana basal, por ejemplo en la amiloidosis, por la extensión de la sustancia amiloide hacia el lado epitelial.
Estadio III. Membrana basal engrosada con incorporación a la misma de los depósitos electrón densos, por encinturamiento de los mismos por las proyecciones membranoides.
Estadio IV. Membrana basal engrosada con variable densidad de los depósitos electrón densos, que le confieren un aspecto agujereado (fig. 10), lesión visible con el tricrómico de Masson en microscopia óptica.
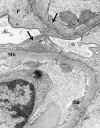
Fig. 8. Glomerulopatía membranosa
estadio I. Lesión incipiente que puede pasar desapercibida, por microscopia
óptica, con glomérulos ópticamente "normales".
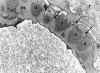
Fig. 9. Glomerulopatía membranosa
estadio II-III. Flechas: proyecciones membranoides de la membrana basal.

Fig. 10. Glomerulopatía membranosa
estadio IV. Aspecto agujereado de la membrana basal.
Con frecuencia los depósitos no son claramente visibles con la tinción tricrómica de Masson, y la única forma de no pasar por alto una glomerulopatía membranosa incipiente es disponer al menos del estudio por microscopía de inmunofluorescencia o microscopia electrónica. Lo mismo se puede aplicar en el despistaje de las amiloidosis incipientes, por lo que ante toda biopsia con diagnóstico clínico de síndrome nefrótico, sobre todo en el adulto, se debe disponer como mínimo de al menos de uno de los dos métodos técnicos. Una buena tinción de plata metenamina es muy útil para apreciar las incipientes rugosidades del lado epitelial de la membrana basal, del estadio I, lesión claramente visible bajo la observación con el objetivo de inmersión. La presencia de depósitos electrón densos en las áreas mesangiales y subendoteliales sugiere una glomerulopatía lúpica, lo que obliga realizar un estudio inmunológico en este sentido, que puede ser corroborado además con la inmunofluorescencia por la positividad para C’1q y C’4.
4. Glomerulopatías con depósitos mesangiales («Glomerulonefritis mesangial IgA. Enfermedad de Berger». «Glomerulonefritis por depósitos mesangiales de IgG»)
La nefropatía por depósitos de IgA (22) fue individualizada merced a la aplicación de la inmunofluorescencia en 1968 por Berger e Hinglais, aunque previamente fue descrita como lesión glomerular por depósitos fibrinoides en los espacios intercapilares por P. Galle y J. Berger (3). La púrpura de Shönlein Henoch muestra cambios similares a nivel renal consistentes en la presencia de depósitos predominantes de IgA en las áreas mesangiales. Son múltiples las causas que se asocian con depósitos de IgA (neoplásicas, infecciones, dermatológicas, cirrosis, etc), lo que obliga a hacer una correlación clínico patológica correcta.
Ultraestructuralmente se evidencia la existencia de depósitos electrón densos a nivel mesangial (figs. 11 y 12); incluso en algunos casos hay extensión subendotelial y/o paramesangial. Se acompaña de aumento de células mesangiales así como expansión de la matriz mesangial. La extensión de la lesión depende del grado de evolución, suficientemente definida por microscopia óptica, estadiándola en cinco fases, que van desde la lesión mínima a la forma avanzada y crónica (14).
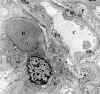
Fig. 11. Glomerulonefritis mesangial
Iga. Depósitos electrón denso de localización mesangial.
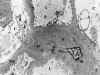
Fig. 12. Transplante renal, recidiva
de nefropatía IgA. Depósitos electrón denso de localizacioón mesangial.
Estadio I. Lesión óptica mínima. Si no se dispone de inmunofluorescencia puede pasar infravalorada y catalogarse la biopsia como normal.
Estadio II. Afectación mínima con pequeñas lesiones focales segmentarias.
Estadio III. Afectación glomerular con lesiones focales segmentarias presentes en menos del 50% de los glomérulos.
Estadio IV. Lesión proliferativa difusa. En algunos caso se asocia con proliferación extracapilar, signo indicativo de mal pronóstico.
Estadio V. Afectación extensa con esclerosis glomerular, atrofia tubular y focos de fibrosis intersticial.
5. Glomerulonefritis proliferativa endocapilar aguda («Glomerulonefritis proliferativa exudativa». «Glomerulonefritis aguda postinfecciosa»)
Proceso glomerular, de curso clínico agudo que se produce por varias causas, siendo las más importantes las postinfecciosas, generalmente bacterianas. Entre las postinfecciosas tienen relevancia las postestrepctocócicas (estreptococo ß-hemolítico grupo A) y se consideran como prototipo de causa de glomerulonefritis proliferativa endocapilar. Se caracteriza ultraestructuralmente por la presencia de gruesos depósitos electrón densos, prototipo de «depósitos inmunológicos», localizados en la vertiente epitelial de la membrana basal, en forma de «jorobas» («humps») (9,14), que corresponden a depósitos de inmunocomplejos (fig. 13). Pueden encontrarse depósitos en otros lugares, tales como subendotelio, intramembranosos y mesangiales.
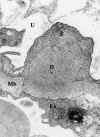
Fig. 13. Glomerulonefritis aguda.
Típica "joroba", depósito electrón denso subepitelial.
Hay varios patrones, imágenes sobre todo catalogadas por el patrón ofrecido mediante inmunofluorescencia; en guirnalda, en cielo estrellado y mesangial (23-26). El patrón mesangial corresponde generalmente a la fase de curación de la enfermedad, y se caracteriza por la presencia de escasos depósitos de localización mesangial. El patrón en cielo estrellado se debe a la presencia de pequeños depósitos, tanto en la pared capilar como a nivel mesangial. El patrón en guirnalda se ve con más frecuencia en varones y en pacientes que presentan proteinuria intensa, y puede ser un indicativo de evolución más crónica y de peor pronóstico a largo plazo (fig. 14). Se observa en un 25% de las biopsias renales y se caracteriza por un depósito denso y prominente subepitelial de inmunoglobulinas y de complemento, que confluye en áreas, siendo los depósitos subendoteliales, intramembranosos y mesangiales menos prominentes. Este patrón confiere a menudo una apariencia lobular al glomérulo, aunque ocasionalmente puede asociarse a lesión membranosa (27). Alguna vez hay dificultad en diferenciar el patrón en guirnalda de una lesión membranosa (5). En el patrón en guirnalda los depósitos subepiteliales tienden a ser más irregulares, y más desiguales en tamaño y distribuidos de forma discontinua en la luz y parcheados en el glomérulo, por depósitos más densos y confluentes, y relacionados con un peor pronóstico de la enfermedad.
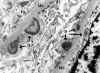
Fig. 14. Glomerulonefritis aguda,
patrón de inmunofluorescencia en guirnalda (IgG, C'3). Numerosos depósitos
electrón densos en forma de jorobas, lesión perfectamente definida en material
recuperado en bloque de parafina. Se observan algún depósito subendotelial.
Otros hallazgos son la presencia de leucocitos neutrófilos adheridos a la membrana basal, que aparece denudada, y pérdida de la continuidad del endotelio de los capilares glomerulares. Membrana basal por lo general normal, tanto en grosor como en contorno a excepción de complicaciones, y asociación con un componente proliferativo extracapilar. La glomerulonefritis aguda puede aparecer en el seno de otras enfermedades glomerulares, como por ejemplo en una glomerulosclerosis nodular diabética de Kimmstiel Wilson (fig. 15) (29), que puede plantear diagnóstico diferencial con la glomeruloenfritis mesangio capilar por el aspecto lobulado e hipercelular de los ovillos.
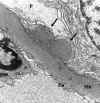
Fig. 15. Glomerulonefritis aguda en
paciente con nefropatía diabética. Membrana basal engrosada. Flechas
depósitos subepiteliales.
6. Glomerulonefritis proliferativa extracapilar («Glomerulonefritis rápidamente progresiva», «Glomerulonefritis necrotizante con semilunas»)
El concepto de glomerulonefritis con semilunas es un término morfológico para designar a un grupo de procesos glomerulares que se caracterizan por la presencia de células en el espacio de Bowman y fibrina, dando lugar a una imagen en semilunas. Se reserva el término de glomerulonefritis difusa extracapilar cuando están afectos al menos un 30% de los glomérulos, correspondiendo al síndrome clínico catalogado como glomerulonefritis rápidamente progresiva, subaguda o maligna, por su evolución a la insuficiencia renal en un periodo corto de tiempo, existiendo una relación pronóstica con el porcentaje de semilunas (habitualmente con muy mal pronóstico cuando la afectación es superior al 50%) (9,13,14).
Hay que señalar que en cualquier forma de glomerulonefritis pueden mostrarse en algún glomérulo, focos de proliferación extracapilar, lo que no hay que catalogar como proliferativa extracapilar difusa; su presencia implica un peor pronóstico. Entre las glomerulonefritis en las que con más frecuencia pueden aparecer la proliferación extracapilar, tenemos la nefropatía de IgA, la agudas postinfecciosas, la púrpura de Shönlein-Henoch, la nefropatía lúpica y algunas glomerulonefritis mesangiocapilares, sobre todo la de por depósitos densos.
Para comprender los hallazgos ultraestructurales es obligado hacer una clasificación de los mismos en base a los hallazgos de la inmunofluorescencia, por mostrar ésta con bastante exactitud el mecanismo patogénico de la lesión.
I. Enfermedad por anticuerpos antimembrana basal (glomerulonefritis rápidamente progresiva tipo I), se caracterizada por una imagen lineal de la membrana basal al aplicar los sueros fluorescentes anti-IgG y anti-C’3. Básicamente hay dos formas; la idiopática y el síndrome de Goodpasture con afectación pulmonar.
II. Enfermedad mediada por inmunocomplejos (glomerulonefritis rápidamente progresiva tipo II), con imagen por inmunofluorescencia de tipo granular o en grumos irregulares, a lo largo de la pared de los capilares glomerulares, con los inmunosueros anti-IgG y anti-C’3, fundamentalmente. La imagen es característica del tipo de inmunocomplejos, por lo que aparece en las nefropatías por inmunocomplejos, como complicación de una aguda postinfecciosa, nefropatía lúpica, nefropatía de IgA, púrpura de Shönlein Henoch. En algunos no se conoce la causa subyacente.
III. Glomerunefritis pauciinmunitaria con semilunas (glomerunefritis rápidamente progresiva tipo III) , caracterizada por mostrar mediante inmunofluorescencia ausencia de fijación tanto lineal como granular al aplicar los inmunosueros antiinmunoglobulinas y complemento. En ocasiones hay escasos depósitos granulares. Se asocian con frecuencia con anticuerpos citoplásmico antineutrófilos (ANCAs) y las vasculitis tipo Wegener y panarteritis micróscopica. En algunas formas de glomerunefritis con semilunas pauciinmunotaria no hay evidencia de vasculitis, por lo que deben ser consideradas como idiopáticas, y con frecuencia son c-ANCA o p-ANCA positivos (30-32).
Se desprende de lo anterior, que los hallazgos ultraestructurales son concordantes con la inmunofluorescencia.
Es común la presencia de fibrina polimerizada en el espacio urinario entre las semilunas (fig. 16) y los ovillos.
Ausencia de depósitos electrón densos, en la formas por anticuerpos antimembrana basal (fig. 17).
Presencia de depósitos electrón densos, tipo depósitos por inmunocomplejos, en las formas con inmunofluorescencia granular (fig. 18). La distribución de los mismos depende del mecanismo de la enfermedad primaria (glomerulonefritis aguda postinfecciosa, lupus...).
En la forma con semilunas pauciinmunitaria, están habitualmente ausentes los depósitos electrón densos de tipo inmunológico.
Hallazgos comunes que varían según el grado de severidad de la lesión son:
Colapso y necrosis de los capilares glomerulares.
Aumento de células en el espacio urinario, a expensas de células epiteliales viscerales y macrófagos.
Rupturas de la membrana basal. Ensanchamiento de la lámina rara externa e hinchazón de las células endoteliales.
Fibras de colágeno cuando hay ruptura de la cápsula de Bowman.
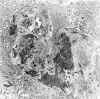
Fig. 16.
Glomerulonefritis proliferativa extracapilar.
Flechas: fibrina.
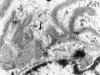
Fig. 17.
Glomerulonefritis extracapilar, tipo por
anticuerpos anti-membrana basal, inmunofluorescencia fijación lineal (IgG).
Ruptura de la membrana basal -> flecha.
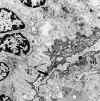
Fig. 18.
Glomerulonefritis extracapilar tipo por
"inmunocomplejos". Flecha depósito subepitelial. Membrana basal
tortuosa.
7. Glomerulonefritis mesangiocapilar («Glomerulonefritis membranoproliferativa», «glomerulonefritis lobular crónica», «glomerulonefritis hipocomplementémica»)
En este grupo están englobadas, básicamente dos formas, la glomerulonefritis mesangiocapilar tipo I, por depósitos subendoteliales, y la glomerulonefritis mesangiocapilar tipo II, por depósitos densos.
La glomerulonefritis membrano proliferativa tipo I, se caracteriza ultraestructuralmente por la extensión circunferencial del mesangio y de la matriz, entre la membrana basal y en endotelio de los capilares glomerulares, con formación de material membranoide a modo de una nueva membrana basal bajo el endotelio, originando la típica imagen de doble contorno de los capilares glomerulares (fig. 19), claramente visible por microscopia óptica sobre todo con las técnicas argénticas. En el espacio subendotelial, subyacente a la membrana basal propia del capilar, se evidencian depósitos electrón densos (fig. 20), de tamaño variable, que cuando son masivos da lugar a una imagen en asa de alambre, lesión característica, pero no específica, de la nefropatía lúpica. También se pueden evidenciar depósitos a nivel mesangial, e incluso subepiteliales (9,14).
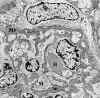
Fig. 19. Glomerulonefritis mesangio
capilar tipo I. En el centro hematíe (H) alrededor del mismo extensión
circunferencial del mesangio.
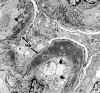
Fig. 20. Glomerulonefritis mesangio
capilar tipo I. Abundantes depósitos electrón densos subendoteliales,
subyacentes a la membrana basal del capilar.
8. Glomerulonefritis por depósitos densos intramembranosos («Glomerulonefritis membrano proliferativa tipo II»)
Aunque existe una similitud clinica con la forma anterior, en nuestra opinión, las carácterísticas morfológicas a nivel histológico, inmunopatológico y ultraestructural, justifican su segregación como forma independiente.
La enfermedad por depósitos densos se caracteriza por la presencia de un depósito electrón de alta densidad, continuo o discontinuo por interrupciones, en la lámina densa de la membrana basal (fig. 21) dividiéndola en dos capas. También están presentes a nivel mesangial, cápsula de Bowman, incluso a nivel de la membrana basal tubular, con cierta polarización hacia el lado intersticial (fig. 22).
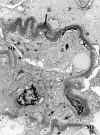
Fig. 21. Enfermedad por depósitos
densos intramembranosos (D flecha), con proliferación extracapilar 100%.
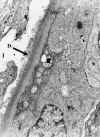
Fig. 22. Mismo caso anterior,
depósitos densos en membrana basal tubular con polarización hacia el lado
intersticial (flecha).
9. Enfermedades con base genética
a. Nefropatía de Alport
Las nefropatías hereditarias son un grupo heterógeneo de afecciones renales de la infancia, unas con un amplio espectro que engloba al sindrome de Alport (33,34) y la hematuria benigna familiar (HFB), todas ellas con alteraciones estructurales de la membrana basal. La nefropatía de Alport es una nefropatía de carácter hereditario que se caracteriza por una anomalía del colágeno tipo IV de la membrana basal. En la forma clásica es una nefropatía evolutiva con insuficiencia renal progresiva que termina en fracaso renal, asociada a sordera neurosensorial y alteraciones oculares, fundamentalmente del cristalino. En la mayoría de los casos se debe a una mutación del gen para la cadena a5 del colágeno IV, COL4A5, que se encuentra en el cromosoma X, (síndrome de Alport ligado al sexo). Otras formas, menos frecuentes, son el síndrome de Alport recesivo y el síndrome de Alport autosómico dominante El colágeno tipo IV es un componente colágeno importante de las membranas basales, y se han secuenciado las 6 cadenas distintas (a1, a2, a3, a4, a5, y a6 y los genes que las codifican (COL4A1, COL4A2, COL4A3, COL4A4, COL4A5, COL4A6). En el cromosoma 13 se encuentran los genes COL4A1 y COL4A2; en el cromosoma 2 se encuentran los genes COL4A3 y COL4A4 y en el cromosoma X los genes COL4A5 y COL6, siendo las delecciones o mutaciones las causantes de la forma clásica del Síndrome de Alport (35).
El estudio ultraestructural, aunque importante, complementa a los estudios genéticos moleculares y de inmunohistoquímica con anticuerpos monoclonales contra tres cadenas del colágeno IV (a1, a3 y a5). Muestra una serie de alteraciones, que aunque características del síndrome de Alport, no deben ser consideradas aisladamente como específicas: 1) Hendiduras de la membrana basal (lámina densa) con aspecto laminado reticulado que encierra áreas electrón lúcidas (fig. 23), 2) Variación del espesor de la membrana basal con zonas de adelgazamiento, semejante a la descrita en la hematuria familiar benigna, 3) Expansión lúcida de la lámina rara interna. Estas alteraciones estructurales también pueden verse en la membrana basal de la cápsula de Bowman y membrana basal tubular. Morfológicamente se han establecido grados de severidad de la lesión renal, tanto en la microscopía óptica (36,37) como por microscopía electrónica (38), Hay estudios morfométricos en un intento de correlacionar los hallazgos morfológicos con el aclaramiento de creatinina y creatinina sérica (39).
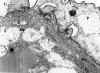
Fig. 23. Nefropatía de Aiport.
Aspecto laminado de la membrana basal.
b. Hematuria familiar benigna
La hematuria familiar benigna, caracterizada por microhematuria permanente con antecedentes familiares, sin insuficiencia renal ni sordera, muestra al estudio ultraestructural un extenso adelgazamiento del espesor de la membrana basal (fig. 24), con valores que van de 225 a 241 nm (39,40). Se puede observar incluso ruptura de la membrana basal con aposición de un podocito (fig. 25). En los grupos de más edad puede haber aumento de matriz y células mesangiales. Hay un aumento de la incidencia en estos pacientes de glomeruloesclerosis focal global cuando se compara con controles como la nefropatía de IgA (36,37).
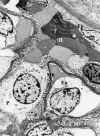
Fig. 24. Hematuria familiar. Numerosos
hematíes en el espacio urinario.
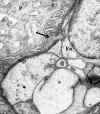
Fig. 25. Hematuria familiar. Membrana
basal adelgazada y ruptura de la misma (flecha).
Las hematurias no familiares que se presentan en niños sin historia familiar de sordera ni de nefropatía, son probablemente por mutación genética de novo (36,37).
En niños con hematuria con / sin sordera neurógena, se debe investigar en la familia la existencia de miembros con insuficiencia renal, hematuria o sordera, y el síndrome de Alport debe estar incluido en al diagnóstico diferencial. Ante la sospecha clínica de nefropatía de Alport o de hematuria familiar benigna, se debe tomar muestra para estudio ultraestuctural, que además nos permite destacar otros procesos que pueden cursar con hematuria, sobre todo con la glomerulonefritis mesangial de IgA.
c. Enfermedad de Fabry
La enfermedad de Fabry es una deficiencia hereditaria recesiva ligada al sexo, del enzima lisosómico a-galactosidasa A (3). El depósito lisosomal corresponde a trihexósido de ceramida (globotriaosilceramida) .
La lesión renal se manifiesta en el aspecto espumoso sobre todo de las células epiteliales viscerales glomerulares, endoteliales, mesangiales, túbulos contorneados distales, íntima de los vasos y arterias de pequeño calibre. En el material incluido en una resina sintética y teñido con azul de metileno, se visualizan densas inclusiones en las células afectas. El estudio ultraestructural renal muestra la presencia de cuerpos de inclusión, redondos u ovalados, de tamaño variable, localizados en todas las células renales, como figuras de mielina, cuyas láminas muestran una periocidad de 40 Å, cuerpos de cebra y estructuras laminadas (fig. 26).
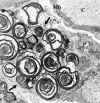
Fig. 26. Enfermedad de Fabry. Podocito
con depósitos osmiófilos constituidos por láminas concéntricas. Flechas
depósitos osmiófilos en forma de cuerpos de cebra.
Abreviaturas usadas en las micrografías electrónicas
B - Cápsula de Bowman
C - Luz capilar
D - Depósito electrón denso
En - Célula endotelial
Ep - Célula epitelial parietal
H - Hematíe
I - Intersticio
L - Leucocitos
l - Lámina densa
M - Célula mesangial
m - Matriz mesangial
Mb - Membrana basal
P - Célula epitelial visceral (podocito)
PB - Célula epitelial parietal
p - Pedicelos
T - Célula tubular
U - Espacio urinario
v - Microvellosidades
BIBLIOGRAFÍA
Oliva H, Navarro B, editores. Patología del riñón. Colección Noticias Médicas, Publicaciones Controladas, S.A. Madrid, 1968.
Barat A. Estudio inmunológico de las nefropatías humanas y su correlación estructural con el microscopio electrónico. Tesis Doctoral. Universidad Autónoma de Madrid, Madrid, 1973.
Oliva H, Barat A, Hernando L, editores. Patología del glomérulo renal. Atlas de microscopia óptica, electrónica e inmunofluorescencia. Salvat Editores, S.A. Barcelona, 1975.
Barat A, Oliva H. Tema monográfico «Glomerulonefritis», JANO Medicina y Humanidades, 1977; 272: 26-38.
Oliva H, Sarasa JL, Rivas C, Barat A, Renedo G, Rivas F, Cortés J. Introducción a la microscopia electrónica diagnostica (I). JANO Medicina y Humanidades 1984; 616: 25-93.
Oliva H, Sarasa JL, Rivas C, Barat A, Renedo G, Rivas F, Cortés J. Introducción la microscopia electrónica diagnóstica (II). JANO Medicina y Humanidades 1984; 618: 27-54.
Barat A, Oliva H. Anatomía patológica de las principales formas de glomerulonefritis. Tema monográfico «Glomerulonefritis», JANO Medicina y Humanidades 1986; 30: 769-88.
Spargo BH. Practical use of electron microscopy for the diagnosis of glomerular disease. Human Pathology 1975; 6: 405-20.
Kern W, Laszik Z.G, Nadasdy T, Silva FG, Bane B.L, Pitha JV, editores. Atlas of renal pathology. W B Saunders Company, 1999, Philadelphia.
Heptinstall RH, editor. Pathology of the Kidney, Vol I, Fourth Edition. Little, Brown and Company, Boston, 1992.
Heptinstall RH, editor. Pathology of the Kidney, Vol II, Fourth Edition. Little, Brown and Company, Boston, 1992.
Heptinstall RH, editor. Pathology of the Kidney, Vol III, Fourth Edition. Little, Brown and Company, Boston, 1992.
Spargo BH, Seymour AE, Ordoñez NG, editores. Renal Biopsy Pathology with Diagnostic and Therapeutic Implications. W Wiley Medical Publication John Wiley & Sons, New York 1980.
Striker G, Striker L, D’Agati VW, editores. The Renal Biopsy. Major problems in pathology. B Saunders, Philadelphia, 1997.
Ruilope LM, Casado S, López de Novales E, Barat A, Hernando L. Síndrome nefrosico infantil con lesiones glomerulares mínimas. Evolucion clinica. Resultados terapeuticos de 40 casos. Rev Clin Esp 1973; 131: 313-8.
Peces R, Sánchez L, Gorostidi M, Álvarez J. Minimal change nephrotic síndrome associated with Hodgkin’s lymphoma. Nephrol Dial Transplant 1991; 6: 155-8.
Korzets Z, Golan E, Manor Y, Schneider M, Bernheim J. Spontannneously remitting minimal change nephropathy proceding a relapse of Hodgkin disease by 19 months. Clin Nephrol 1992; 38.
Rich AR. A hitherto indescribed vulnerability of the juxtamedullary glomeruli in the lipoid nephrosis. Johns Hopkins Med J, 1957; 100: 173.
Cannata J, Pérez R, Egido J, Barat A, Hernando L. Glomerulonefritis membranosa idiopatica. evolucion clinica e histologica. Rev Clin Esp 1978; 151: 371-5.
Plaza, Herrero G, Barat A, Loutaif L, Hernando L, Vallado P, Oliva H. Membranous glomerulonephritis as a complication of oral gold therapy. J.J. Annals of Internal Medicine 1982; 97: 563.
Ehrenreich T, Churg J. Pathology of membranous nephropathy. En Kidney Pathology Decenial 1966-1975, Series Editor, Sheldon C. Sommers, Apleton-Century-Crofts. New York, 1975, p. 389-433.
Rivera F, Egido J, Alvarez V, Barat A, Hernando L. Glomerulonefritis mesangial IgA. aspectos clinicos y evolutivos de ochenta casos. Rev Clin Esp 1982; 164: 41-8.
Edelstein CL, Bates WD. Subtypes of acute postinfectious glomerulonephritis: a clinico-pathological correlation. Clin Nephrol 1992; 38: 311.
Melbey PC, Musick WD, Luger AM, Khanna R. Poststreptococcal glomerulonephritis in the elderly: Report of a case and review of the literature. Am J Nephrol 1987; 7: 235.
Hinglais N, García-Torres R, Kleinknecht D. Long-term prognosis in acute glomerulonephritis: The predictive value of early clinical and pathologic features observed in 65 patients. Am J Med 1974; 56: 52.
Mclean RH, Schranger MA, Rothfield NF, Berman MM. Normal complement in early poststreptococcal glomerulonephritis. Br Med J. 1977; 1: 1326.
Richet G, Fillastre JP, Morel-Maroger L, Bariety J. Change from diffuse proliferative to membranous glomerulonephritis. Serial biopsies in four cases. Kidney Int 1974; 5: 57.
Sorger K, Gessler M, Hubner FK, Kohler H, Olbing H, Schulz W, Thoenes GH, Thoenes W. Follow-up studies of three subtypes of acute postinfectious glomerulonephritis ascertained by renal biopsy. Clin Nephol 1987; 27: 111.
Plaza JJ, Cortes J, Rivera F, Peces R, Barat A, Hernando L, Oliva H. Glomerulonefritis aguda postestreptococica complicando a una nefropatia diabetica. Rev Clin Esp 1981; 162: 49-51.
Jennette JC, Falk RJ. Small-vessel vasculitis [Review]. N Engl J Med 1997; 337: 1512-23.
Matteson EL, Jennette JC, Fak RJ. SmaIl-Vessel Vasculitis. N EngI J Med 1998; 338: 994-995, Apr 2, 1998.
Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with speciflcity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulone phrits [Abstract]. N EngI J Med 1988; 318: 1651-7.
Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J 1927; 1: 504.
PerkotT GT, Stephens FE, Dolowitz DA, Tyler FH. A clinical study of hereditary interstitial pyelonephritis. Arch Intem Med 1951 ;88: 191.
Lemmink llli, Nillesen WN, Mochizuki T, Schroder CH, Brunner
HG, van Oost BA, Monnens LA, Smeets HJ. Benign familial hematuria due to
mutation of the type IV collagen alpha4 gene. J Clin Invest 1996; 98:
1114-8.
Bailey RR. Familial haematuria due a thin basement membrane nephropathy. N Z
Med J, 1990; 103: 312-3.
Grünfeld JP. The clinical spectrum of hereditary nephritis. Kidney Intemational 1985; 27: 83-92.
Gubler G, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, Habib R. Alpot’s Syndrome. A report of58 cases and a review of the literature. Am J Med 1981; 70: 493- 505.
Coleman M, Haynes WDS, Dimopoulos P, Barrat LJ, Jarvis LR. Glomerular base- ment membrane abnormalities associated with apparently idiopathic hematuria: ultras- tructural morphometric analysis. Hum Pathol 1986; 17: 1022-30.
Fierro R, Alegre R, Barat A, Hemando L, Casado S, Oliva H. Enfermedad de Alport y Hematuria Recidivante: Estudio comparativo y revisión de la literatura. Patología (Mex) 1994; 32: 21-7.
![]()