
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
Socorro María Rodríguez-Pinilla, Amparo Benito-Berlinches, Claudio Ballestin, Gabriel Usera
Departamento de Anatomía Patológica. Hospital Universitario 12 de Octubre. Madrid.
SUMMARY
We report a case of primary angiosarcoma of the adrenal gland with a review of the literature in order to discuss its clinical-pathological features. With only 21 previously reported cases it is an uncommon entity whose proper diagnosis is difficult to make. Clinically it can be confused with benign cystic processes, especially if it takes an indolent course. Microscopically, its unexplained tendency toward epithelioid characteristics include it in the differential diagnosis with the more common epithelial neoplasms. Its etiological factors are completely unknown and despite being biologically malignant neoplasms it seems to have a relatively good prognosis.
Key words: Angiosarcoma, malignant vascular neoplasms, epithelioid, adrenal gland.
RESUMEN
Presentamos un nuevo caso de angiosarcoma de suprarrenal y revisamos la literatura con la intención de exponer las características clínico-patológicas de esta entidad. Con tan sólo 21 casos descritos, es una entidad muy poco frecuente y de difícil diagnóstico. Clínicamente pueden confundirse con procesos quísticos benignos, especialmente si cursan de forma asintomática. Microscópicamente y por razones desconocidas tienden a presentan morfología epitelioide, lo que los incluye en el diagnóstico diferencial de neoplasias epiteliales mucho más frecuentes. Los factores etiológicos se desconocen por completo y aúnque biológicamente se trate de neoplasias malignas parecen tener sin embargo un pronóstico relativamente bueno.
Palabras clave: Angiosarcoma, neoplasia vascular maligna, epitelioide, suprarrenal.
INTRODUCTION
Primary mesenchymal neoplasms of the adrenal gland are rare, and a malignant one is an extraordinary finding. Published examples include: leiomyosarcomas, malignant peripheral nerve sheath tumor and angiosarcomas (1). Malignant vascular neoplasms within the adrenal gland have a singular appearance, both macroscopically and microscopically. For this reason, and due to their rarity, they can easily be misdiagnosed, not only by clinicians but also by pathologists. We present a new case and a review of the literature in an attempt to better define the clinicopathologic features of these neoplasms and their biologic potential.
CLINICAL HISTORY
A 61-year-old man presented in our hospital complaining of an intense pain of two days duration in the right upper abdomen radiating to the lumbar area. He also referred a three-months constitutional syndrome with a 7 kilogram weight loss. Complete blood count showed leukocytosis and anemia. He worked as a carpenter and smoked two packs of cigarettes per day, but had no other medical history. Computed tomography of the abdomen revealed a sharply-delimited space- occupying mass extending from the unaffected right lobule of the liver to the upper pole of the kidney, destroying its inner contour line. It measured 12 cm at its greatest diameter and had several densities inside (fig. 1). Further aortographic studies revealed a non contrast-enhancing formation. All these related properties suggested it was a complicated renal cyst. An «en block» resection of the right kidney was performed and an amorphous hemorrhagic mass attached to it removed. Under light microscopy and supported by immunohistochemical studies, diagnosis was made of epithelioid variant of angiosarcoma of the adrenal gland. There was expansion of the neoplastic cells beyond the adrenal gland capsule into the periadrenal fat tissue with no renal invasion.
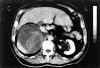
Fig. 1. Tomographic studies revealed a non contrast-enhancing formation
destroying the upper pole of the right kidney.
In an attempt to exclude another primary origin of this process several radiographic studies were performed. No masses in thorax, mediastinum, retroperitoneum or abdomen were found. There was no splenomegaly, hepatomegaly nor adenopathies.
After surgical removal of the tumor, adjunctive chemotherapy or radiotherapy was not considered because it was believed that the entire tumor had been removed. To date (three years later), the patient is well and free of tumor.
MATERIAL AND METHODS
The surgical specimen received was fixed in 6% formalin. Part of the tissue was embedded in paraffin and cut into sections of 5 m thick that were stained with hematoxylin and eosin. In addition, immunohistochemical studies to cytokeratin (AE1-AE3, 1:50, Dako), EMA (E29, 1:100, Dako), desmin (D33, 1:100, Dako), vimentin (V9, 1:1000, Dako), smooth muscle actin (1 A 4, 1:50, Dako), Factor VIII antigen (polyclonal, 1:500, Dako), CD31 (JC/70 A, 1:20, Dako), Collagen IV (CIV 22, 1:100, Dako) and S-100 protein (polyclonal, 1:2000, Dako) were performed on paraffin sections using the ABC (avidin-biotin peroxidase complex).
PATHOLOGY AND LABORATORY FINDINGS
Gross findings
Gross pathologic examination of the surgical specimen, revealed a morphologically normal kidney that measured 8 ¥ 6 ¥ 4 cm. A brown, shattered mass of 12 cm in greatest diameter that partly infiltrated the perinephric fat was attached to its upper pole. It was almost exclusively cystic and had pieces of this fleshy tissue mixed with a slightly coagulated hemorrhagic liquid inside. An elongated orangey layer was found on the lower part of this mass while it was being cut into slices, and suggested a possible adrenal origin.
Histopathological findings
Microscopically, we found a cellular proliferation that effaced the normal appearance of the adrenal gland. The mass consisted of tubular, pseudoglandular or alveolar structures that were intermixed with solid foci of morphologically tumor cells (fig. 2a). These cells had a pleomorphic appearance, alternating from cuboidal to quadrangular. The round-to-oval nuclei with a conspicuous eosinophilic nucleolus were almost always centrally placed. When they were arranged in tubules, there was a single row of lining cells that occasionally coalesced to form cell nests projecting into the lumen. Red blood cells could frequently be identified in the lumen suggesting that they were vascular spaces. Moreover, a few vesicular cytoplasmic formations with the nuclei toward the periphery of the cell were also seen from time to time. These also contained red blood cells and simulated primitive capillaries (fig. 2b).
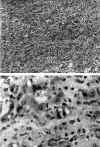
Fig. 2. Tumoral proliferation of
cells arranged in tubules or in solid foci. Hematoxilin-eosin stain x100. B.
Tumoral cells that had vesicular cytoplasmic formations with red blood cells
inside, simulating primitive capillaries. Hematoxilin-eosin stain x 400.
The presence of a huge univacuolar cystic formation that was lined by the previously described cells was also found. The adrenal cortex formed part of its wall and clotted hemorrhagic material was observed in its luminal space (fig. 3).
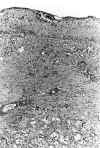
Fig. 3. Normal adrenal cortex and
part of the wall of a hugh neoplastic cyst. Hematoxilin-eosin x100.
The stroma consisted mainly of thin fibrovascular connective tissue associated with sparse inflammatory cell infiltrate. Ferric foamy macrophages were also seen. Reticular stains enhanced vascular spaces without delimiting each single cell in the solid parts. No more than 20 per cent of normal adrenal cortex tissue remained and this was identified intermixed in the tumoral mass as single cells or clusters among the neoplastic cells. Some of normal cells were compressed while others were plump with a highly hyperchromatic nucleus.
Immunohistochemical findings
Immunohistochemical stains showed intense positivity for vimentin and CD31. This reactivity was seen in the cells lining the luminal vascular spaces as well as in the solid areas, confirming its endothelial origin. There was no immunoreactivity for the rest of the antibodies used in this study. Collagen type IV antigen was expressed surrounding vascular spaces.
DISCUSSION
Primary angiosarcomas of the adrenal gland are rare malignant neoplasms of difficult diagnosis. To the best of our knowledge, there are only 21 previously reported cases. The first case was described in 1988 by Kareti et al (2). The largest study was done at the Armed Forces Institute of Pathology by Wenig et al where 9 cases (including the first case), were described (3). The other reports are single cases (4-14).
The etiological factors of this entity remain unknown. Except for a case reported in a vineyard worker exposed to arsenic containing insecticides for over 20 years (5), and another that was associated with an abdominal fibromatosis (6), none of the reported cases, including ours, had any available carcinogenic exposure that could potentially be linked to the development of angiosarcoma. None of the cases, including ours, have any family history of adrenal neoplasms suggestive of a MEN syndrome, a prior history of abdominal radiotherapy or long-term androgenic anabolic steroid treatment.
It occurs most frequently in the sixth and seventh decades of life (age range, 41-85 years), the some median age range as in angiosarcomas in general (15). Men are the most frequently affected. Of the 21 cases reported 12 were men, 6 women and 3 were not specified.
The most commonly reported symptoms have been pain and abdominal mass. Other complaints have been weight loss, fever and weakness. Two cases were asymptomatic, one presented as a paraneoplastic syndrome (4) and another debuted with metastases to bone and liver (8). None of the patients were hypertensive or had a history of Addison’disease.
Grossly, the neoplasms varied from well-circumscribed to invasive. All previously reported cases were solid to cystic in appearance ranging in size from 5 to 10 cm, although our case was an almost exclusively cystic mass that measured 12 cm at its greatest diameter. Aortography revealed a hypovascular mass in most cases.
Microscopically, all cases shared unexplained tendency toward an epithelioid appearance and immunoreactivity for keratins was seen in 12 of the 21 previously reported cases (3,4,6,8,11,14) while only 3 were negative (3,5).
A proper diagnosis of this tumor is difficult to make: The facts that these neoplasms can be well circumscribed and non contrast-enhancing suggest a benign, non-neoplastic process to the clinicians. Its peculiar histologic and immunological features as well as its low incidence induce pathologists to confuse it with adrenal epithelial neoplasms.
Adrenal cortical carcinoma, pheochromocytoma, metastatic (adeno)carcinoma, metastatic malignant melanoma and another primary origin of an angiosarcoma are malignant entities that should always be ruled out. Among the benign neoplasms that may simulate epithelioid angiosarcoma are adrenal adenomas undergoing massive hemorrhage and epithelioid hemangioendothelioma (3).
Immunohistochemical studies are of particular help when most of the tumor follows a solid pattern. A combination of endothelial-related markers (such as CD34, FVIII antigen and CD31) must be used in the antigen panel of these neoplasms, based on their limitations in terms of sensitivity and specificity (3,16,17). Reactivity for cytokeratins, a well-known phenomenon previously mentioned, appears in a wide variety of mesenchymal neoplasms and does not seem to be related to an epithelial histogenetic origin (3,16,18).
In our case several radiographic studies and an exhaustive physical exploration were made to exclude skin, soft tissue or intraparenchymal masses in any other internal organs. Microscopically it matched properly into the epithelioid variant of angiosarcoma (15), was negative for keratins and immunoreacted positively for CD31.
Adrenal angiosarcomas are biologically malignant neoplasms with a capacity not only to infiltrate locally but also to metastasize. Five reported cases metastasized, one to bone and liver (8), one to the pleura (9) and three to the lung (3).
The treatment of patients with adrenal angiosarcoma is still controversial, because of the limited experience with this tumor. After reviewing of the literature we found 12 cases that were exclusively treated by adrenalectomy (3,4,7,9-11) with or without accessory splenectomy (3) or nephrectomy (7); only four were alive and well after 1 (11), 11 (3) and 13 (3) years respectively. The follow-up period of the fourth (4) was not specified. Another four patients died due to post-operative complications (3,5,9,10) with evidence of disease in just one case (9). Four other patients received only surgical treatment but died with disease (3,7).
In two cases surgical treatment was performed in conjunction with chemotherapy (3): one died without evidence of disease 4 years after surgery and the other was alive with no evidence of disease 6 years after treatment. In one case, radiation therapy was applied following adrenalectomy but no posttreatment results have been published. Three of the cases that metastasized received only surgical treatment: one died postoperatively, the second died with disease 1 year after treatment and the third was alive with no evidence of disease after 11 years follow-up. One case received chemotherapy and died with no evidence of disease 4 years after surgery and there are no records of the case that metastasized to bone and liver. No treatment data are available from the rest of the case reports.
In conclusion, it seems that surgical eradication (with regular and frequent controls) has good outcome in more than half of the cases, despite the biology of this tumor.
Although the present case was quite big in size, it only infiltrated the periadrenal fat focally and had not metastasized. Only surgical treatment was considered and after three years follow-up, our patient is well and free of tumor.
REFERENCES
Rosai J. Adrenal gland and other paraganglia. En: Rosai J, editor. Ackerman´s surgical pathology. 8th ed. St Louis: Mosby; 1996, p. 1043-4.
Kareti LR, Katlein S, Siew S, Blauvelt A. Angiosarcoma of the adrenal gland. Arch Pathol Lab Med 1988; 112: 1163-5.
Wenig BM, Abbondanzo SL, Heffess CS. Epithelioid Angiosarcoma of the adrenal glands. A clinicopathologic study of nine cases with a discussion of the implications of finding «Epithelial-Specific» markers. Am J Surg Pathol 1994; 18: 62-73.
Bosco PJ, Silverman ML, Zinman LM. Primary angiosarcoma of adrenal gland presenting as paraneoplastic syndrome: Case report. J of Urol 1991; 146: 1101-3.
Livaditou A, Alexiou G, Floros D, Filippidis T, Dosios T, Bays D. Epithelioid Angiosarcoma of the adrenal gland associated with cronic arsenic intoxication? Path Res Pract 1991; 187: 284-9.
Ben-Izhak O, Auslander L, Rabinson S, Lichtig C, Sternberg A. Epithelioid angiosarcoma of the adrenal gland with cytokeratin expression. Report of a case with accompanying mesenteric fibromatosis. Cancer 1992; 69: 1808-12.
Fiordelise S, Zangrandi A, Tronci A, Rovereto B, Valentino R.V, Bezzi E. Angiosarcoma of the adrenal gland: Case report. Arch-Ital-Urol-Nefrol-Androl 1992; 64: 341-3.
Jochum W, Schroder S, Risti B, Marincek B, von Hochstetter A. Cytokeratin-positive angiosarcoma of the adrenal gland. Pathologe 1994; 15: 181-186.
Mc Cleary AJ. Massive haemothorax secondary to angiosarcoma. Thorax 1994; 49: 1036-7.
Sasaki R, Tachiki Y, Tsukada T, Miura K, Kato T, Saito K. A case of adrenal angiosarcoma. Nippon-Hinyokika-Gakkai-Zasshi 1995; 86: 1064-7.
Abboud E, Weisenberg E, Khan S, Rhone DP, Chicago. Pathologic quiz case. Arch Pathol Lab Med 1999; 123: 157-8.
Croitoru AG, Klausner AP, Mc Williams G, Unger PD. Primary epithelioid angiosarcoma of the adrenal gland. Ann Diagn Pathol 2001; 5: 300-30.
Ferrozzi F, Tognini G, Bova D, Zuccoli G, Pavone P. Hemangiosarcoma of the adrenal glands: CT findings in two cases. Abdom Imaging 2001; 26: 336-9.
Kruger S, Kujath P, Johannisson R, Feller AC. Primary epithelioid angiosarcoma of the adrenal gland. Case report and review of the literature. Tumori 2001; 87: 262-5.
Weiss SW, Goldblum JR. Malignant vascular tumors. En: Enzinger FM, Weiss SW, editores. Soft tissue tumors. 4th ed. St. Louis: Mosby; 2001. p. 917-54.
Mackay B, Ordoñez N, Huang W. Ultrastructural and Immunocytochemical observations on angiosarcomas. Ultrastructural Pathology 1989; 13: 97-110.
Fletcher C, Beham A, Bekir S, Clarke AM, Marley NJ. Epithelioid angiosarcoma of deep soft tissue: A distinctive tumor readily mistaken for an epithelial neoplasm. Am J Surg Pathol 1991; 15: 915-24.
Gray M, Rosenberg A, Dickersin G, Bhan AK. Cytokeratin Expression in epithelioid vascular neoplasms. Hum Pathol 1990; 21: 212-7.
![]()