
Vol. 36, n.º 4, 2003
|
REVISTA
ESPAÑOLA DE
Vol. 36, n.º 4, 2003 |
María Niveiro de Jaime, F. Ignacio Aranda López, Gloria Peiró Cabrera
Servicio de Patología. Hospital General Universitario de Alicante. niveiro_mar@gva.es
RESUMEN
Los adenomas hipofisarios son neoplasias benignas originadas en células de la adenohipófisis. Representan el tumor más habitual en la silla turca y constituyen un 10-15% de las neoplasias intracraneales. Inicialmente fueron clasificados, según sus características tintoriales, en adenomas acidófilos, basófilos y cromófobos. La aplicación de técnicas morfológicas como la microscopía electrónica y la inmunohistoquímica, y la integración de los hallazgos morfológicos con los datos clínicos, de laboratorio y de las técnicas de imagen han permitido el desarrollo de la nueva clasificación de los adenomas hipofisarios. Las técnicas genéticas y moleculares están proporcionando información que ayuda a entender la patogénesis de algunas variedades de adenoma hipofisario. Por otra parte, la aplicación de determinados marcadores puede aportar información pronóstica y ayudar a predecir la respuesta a modalidades terapéuticas específicas.
Palabras clave: adenoma hipofisario, hipófisis, patología, inmunohistoquímica, microscopía electrónica, proliferación, patología molecular.
SUMMARY
Pituitary adenomas are benign neoplasms originating in adenohypophyseal cells. They are the most common neoplasms found in the sellar region, comprising 10-15 % of all primary intracranial tumors. Based on the tinctorial characteristics of the cell cytoplasm, they have been traditionally classified as acidophilic, basophilic and chromophobic adenomas. The integration of the clinical and laboratory findings with the data obtained by imaging techniques and by morphological techniques (such as electron microscopy and immunohistochemistry) has led to a new pituitary adenoma classification. The development and application of molecular and genetic techniques are providing information about the pathogenesis of pituitary adenomas. Also, specific markers can be of help in prognostic evaluation and in predicting responses to certain therapeutic modalities.
Key words: pituitary adenoma, hypophysis, pathology, immunohistochemistry, electron microscopy, proliferation, molecular pathology.
INTRODUCCIÓN
Los adenomas hipofisarios (AH) son expansiones clonales de células adenohipofisarias que pueden originar una amplia variedad de síndromes clínicos derivados de la producción de una o varias hormonas, o secundarios al crecimiento local. Los AH constituyen un 10-15% de los tumores intracraneales, si bien el estudio cuidadoso de las hipófisis en autopsia muestra su presencia en un 20 % de los casos (1-3). El aumento de la sensibilidad diagnóstica con la incorporación de técnicas como la TAC o la resonancia magnética, ha determinado un aumento de su prevalencia en series clínicas (3,4)
La clasificación anatomopatológica de los AH se basó inicialmente en características tintoriales, que sin embargo presentan pobre correlación con la forma de presentación clínica. La ultraestructura y la aplicación sistemática de la inmunohistoquímica hormonal han permitido el desarrollo de clasificaciones funcionales con significado clínico (1,3,5,6), y permiten encuadrar los AH en tres familias principales (tabla 1). La inmunohistoquímica ha puesto de manifiesto, por otra parte, la alta prevalencia de los adenomas plurihormonales y gonadotropos, que en ocasiones origina problemas de interpretación y clasificación (tabla 2) (3,5). Además de la valoración hormonal, la inmunohistoquímica puede aportar información complementaria que puede no hacer necesario el estudio ultraestructural con fines diagnósticos. Las tinciones con citoqueratinas o con marcadores de proteínas mitocondriales permiten la valoración de aspectos hasta hace poco limitados a los estudios basados en la microscopía electrónica. Además, el estudio de la actividad proliferativa con Ki-67 o de la expresión de determinadas proteínas codificadas por genes tumor supresores como p53 también ha aportado información de interés. Finalmente las aportaciones de las técnicas moleculares pueden ayudar a entender la patogénesis de algunas variedades de adenoma.

El objetivo del siguiente artículo es revisar la patología de los AH, especialmente en lo referente a su valoración inmunohistoquímica, así como de los aspectos moleculares que pueden estar implicados en el desarrollo de los diferentes tipos de AH.
ADENOMAS SOMATOTROPOS
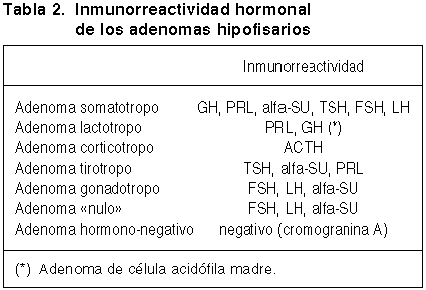
Los adenomas somatotropos, constituyen el 10-15% de todos los adenomas hipofisarios (2,3). La mayoría son tumores funcionantes que segregan hormona del crecimiento (GH) lo que provoca acromegalia en adultos y gigantismo en niños. En el suero se observan niveles elevados de hormona del crecimiento e IGF-1 («insulin growth factor») y, con frecuencia, aumento de otras hormonas como prolactina (PRL) y subunidad alfa (2,3). Radiológicamente, la mayoría (70-80%) son macroadenomas (>10 mm) (fig. 1) (1-3). En ausencia de tumor hipofisario debe descartarse acromegalia secundaria a la secreción ectópica de GH o de GHRH («growth hormone releasing hormone») originada por otros tumores endocrinos como tumores carcinoides (7,8).
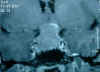
Fig. 1. Imagen de resonancia
magnética nuclear en T1, que muestra un macroadenoma hipofisario (adenoma
somatotropo densamente granulado) con extensión supraselar.
Desde el punto de vista morfológico, con tinciones convencionales estos adenomas son descritos como adenomas acidófilos o cromófobos (fig. 2). La microscopía electrónica permite clasificarlos en densa o pobremente granulados, observándose en algunos casos características mixtas (2,3,9). Los pobremente granulados muestran escasos gránulos de secreción de distribución periférica, y de forma característica, ovillos de filamentos en situación paranuclear (10) (figs. 3A-3B), lo que puede ser observado con microscopía óptica como cuerpos hialinos globoides intracitoplásmicos y que corresponden a citoqueratinas como demuestra la inmunohistoquímica (fig. 4). La inmunohistoquímica hormonal permite determinar la presencia de GH en las células neoplásicas y realizar la estimación de la densidad granular. Los adenomas pobremente granulados presentan menor inmunorreactividad citoplasmática para GH, preferentemente de disposición periférica (fig. 5) y menor proporción de células positivas que los adenomas densamente granulados que muestran positividad granular difusa en todo el citoplasma, presente en la mayoría de las células (3,9,11). Se observa, además, frecuente positividad para prolactina (fig. 6), que en algunas series alcanza la totalidad de los casos (12).
Fig. 2. Adenomas somatotropo
pobremente granulado, tintorialmente cromófobo (H&E x400).

Fig. 3A. Ultraestructura de adenoma
somatotropo pobremente granulado, con escasos gránulos de secreción de
disposición periférica y cuerpo fibroso yuxtanuclear (x9120).

Fig. 3B. Ultraestructura de adenoma
somatotropo pobremente granulado: detalle de cuerpo fibroso, constituido por
filamentos intermedios. En su espesor se identifican centríolos, vesículas de
retículo endoplásmico y gránulos (x25080).
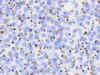
Fig. 4. Adenoma somatotropo
pobremente granulado. Patrón inmunohistoquímico paranuclear "en
gota" con citoqueratina (AE1: AE3, estreptavidina biotina peroxidasa x200).
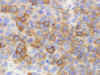
Fig. 5. Adenoma somatotropo
pobremente granulado. Positividad para GH (estreptavidina biotina peroxidasa
x400).
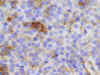
Fig. 6. Adenoma somatotropo
pobremente granulado. Positividad para prolactina (estreptavidina biotina
peroxidasa x400).
El anticuerpo Ki-67 permite estimar la actividad proliferativa y determinar si existen diferencias según el fenotipo morfológico (13). Se ha observado que los adenomas somatotropos localmente agresivos presentan mayor actividad proliferativa con Ki-67 (14). La inmunohistoquímica también permite valorar la presencia de cambio oncocítico en las células del adenoma. Algunos autores, describen un comportamiento más agresivo en adenomas somatotropos que presentan células oncocíticas (15).
Recientemente se ha estudiado el papel de la angiogénesis en los tumores hipofisarios. Los adenomas somatotropos parecen presentar menor densidad en microvasos (DMV) que otros tipos de adenoma. La DMV no parece asociarse con la actividad proliferativa medida con Ki-67 (16).
Las hipótesis más recientes sobre tumorogénesis de los AH sugieren que determinadas alteraciones genéticas juegan un papel en la iniciación y promoción de estas neoplasias. Una alteración génica descrita en los adenomas somatotropos consiste en la mutación en el gen que codifica la subunidad alfa de las proteínas G (gen GNSA1) (17,18). La mutación produce una ganancia de función de las proteínas G, adquiriendo las células mutadas la capacidad de mantener niveles altos de AMPc, independiente de las señales extracelulares (actividad adenilatociclasa constitutiva) que da lugar a la proliferación mantenida de las mismas (17). Las mutaciones hasta ahora descritas se localizan en el codón 201 (exón 8) y codón 227 (exón 9) de dicho gen (19). Esta mutación denominada «gsp» ha sido descrita entre otros tumores, en adenomas somatotropos, con frecuencias según las series, comprendidas entre el 4,4% y 40% (17-22). Varios grupos han intentado determinar las características clínicas, biológicas y morfológicas de los adenomas con mutación gsp. Algunos trabajos han descrito que los adenomas GH gsp+ presentan menor tamaño (17,20,22) y cifras basales menores de GH (17), lo que no es corroborado por otros autores (19). En relación con el fenotipo morfológico, la mutación gsp parece ser más frecuente en adenomas densamente granulados (22), lo que no ha sido confirmado en otros estudios (23). Por último también se ha sugerido que los pacientes con adenomas gsp + presentarían una mayor sensibilidad al tratamiento con análogos de la somatostatina como el octeótrido (20,24), que es el tratamiento médico de elección en estos pacientes.
El POU1F1, descrito inicialmente en familias con déficit de GH, prolactina y TSH es un gen que codifica una proteína de 33 kD conocida como Pit-1/GHF1 (factor de transcripción nuclear específico de la hipófisis), que actúa como promotor de los genes de GH y prolactina (25). La proteína Pit-1 esta expresada en adenomas somatotropos, prolactinomas y tirotropinomas, pero sólo de forma excepcional en otros tipos de adenoma hipofisario (25,26). Los estudios cuantitativos de Pit-1 no han demostrado sobrexpresión en adenomas somatotropos o mamotropos, por lo que este gen no parece jugar ningún papel en la tumorogénesis de este grupo de adenomas (27).
El gen PTTG («pituitary tumor transforming gene»), recientemente identificado, se localiza en el cromosoma 5q33 y codifica una proteína de 202 aminoácidos (28,29). Su función parece estar relacionada con estimulación de factores de crecimiento como bFGF e inducción angiogénesis (30). Zhang y cols observan un aumento de expresión de PTTG en los 13 adenomas somatotropos estudiados (30).
El tratamiento de la acromegalia se basa en la cirugía del tumor hipofisario. En caso de resección incompleta debe realizarse tratamiento con radioterapia. Un procedimiento alternativo a la cirugía es el tratamiento con análogos de somatostatina como el octeótrido (lanreótido) (31). También puede aplicarse de forma preoperatoria, habiéndose documentado disminución del volumen tumoral, con reducción significativa de las cifras de GH, IGF-I e IGFBP-3 («IGF-binding protein») (32).
ADENOMAS ASOCIADOS A HIPERPROLACTINEMIA
Prolactinoma (adenoma lactotropo)
Es el tipo más frecuente de AH, si bien su buena respuesta al tratamiento médico ha determinado que su prevalencia en las series quirúrgicas sea baja. La secreción en exceso de prolactina originada por estos tumores provoca en la mujer el síndrome de amenorrea-galactorrea, mientras que en el hombre puede haber pérdida de la libido e impotencia (3). Las cifras de prolactina suelen estar elevadas por encima de 200 µg/ml. En el prolactinoma las pruebas de estimulación con TRH («TSH-releasing hormone») no modifican las cifras de prolactina. En relación con el tamaño tumoral, los microadenomas suelen presentarse en mujeres jóvenes y en los bordes laterales de los lóbulos de la adenohipófisis, siendo raramente invasivos; los macroadenomas suelen tener un comportamiento localmente destructivo con invasión del seno esfenoidal o extensión supraselar (3). Algunos estudios, sin embargo, no encuentran diferencias en el grado de invasión o en el tamaño tumoral de prolactinomas procedentes de mujeres jóvenes, postmenopáusicas y hombres (33).
En general los prolactinomas son tumores tintorialmente cromófobos, con crecimiento difuso, aunque en ocasiones el citoplasma puede tener una leve basofilia debido a la gran cantidad de retículo endoplásmico rugoso que presentan sus células (1,3). Desde el punto de vista ultraestructural se caracterizan por estar constituidos por células ricas en retículo endoplásmico rugoso, con aparato de Golgi paranuclear bien desarrollado, con gránulos inmaduros (fig. 7). Los gránulos de secreción son, en general, escasos. De manera característica es posible observar gránulos que vierten su contenido entre dos células, lo que ha sido denominado exocitosis de situación anormal (34) (fig. 8). La inmunohistoquímica es característica, con positividad paranuclear para prolactina (patrón denominado tipo Golgi) (fig. 9), si bien pueden expresar otras hormonas hasta en un 50% de los casos (35). También pueden presentar positividad nuclear para receptores estrogénicos y para Pit-1. Mucho más infrecuente es el prolactinoma denso en gránulos (adenoma lactotropo densamente granulado), tintorialmente acidófilo y caracterizado por una mayor densidad en gránulos, que pueden alcanzar los 700 nm de diámetro. En este subtipo la positividad inmunohistoquímica para prolactina es citoplasmática difusa.
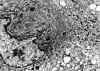
Fig. 7. Prolactinoma pobremente
granulado. Imagen ultraestructural con retículo endoplásmico rugoso prominente
y aparato de Golbi bien desarrollado (x13600).

Fig. 8. Prolactinoma pobremente
granulado. Exocitosis en situación anormal (x47880).

Fig. 9. Inmunorreactividad para
prolactina, con patrón de positividad paranuclear (tipo Golgi) (estreptavidina
biotina peroxidasa x200).
El prolactinoma es el tipo de adenoma más frecuentemente observado en la neoplasia maligna múltiple tipo I (MEN-I). En un 15-20% el tumor hipofisario puede ser la primera manifestación de la enfermedad. El 85 % corresponden a macroadenomas, con extensión extraselar en el 30% (36).
Un hallazgo que puede observarse en el prolactinoma es el depósito de amiloide, que con frecuencia presenta configuración esferoidal y que parece originarse en prolactina inadecuadamente procesada (37).
Adenoma de célula acidófila madre
Esta variedad de prolactinoma, de comportamiento más agresivo, se caracteriza por presentar hiperprolactinemia asociada a síntomas menores derivados de la secreción de GH. La morfología corresponde a un adenoma tintorialmente cromófobo o levemente acidófilo, con arquitectura difusa. La inmunohistoquímica demuestra positividad para prolactina, aunque no con el característico patrón Golgi de los prolactinomas, siendo posible la demostración en una proporción variable de células positivas para GH (38). Con frecuencia acumula mitocondrias, que pueden presentar gran tamaño (megamitocondrias), lo que puede ser la causa de la acidofilia que presentan en ocasiones. Por otra parte, el cambio oncocítico en un prolactinoma puede ser indicativo de mayor agresividad (15). Estos tumores ocasionalmente contienen cuerpos fibrosos, lo que puede ser demostrado mediante tinción para citoqueratinas, observándose positividad en «gota» paranuclear. Las alteraciones mitocondriales observadas en el estudio ultraestructural pueden ayudar en el diagnóstico. Su identificación tiene importancia ya que son adenomas con mayor agresividad local y tendencia a la recidiva. Presentan, además, pobre respuesta al tratamiento con bromocriptina.
Los estudios moleculares realizados en los prolactinomas no aportan información relevante. La mutación gsp no juega ningún papel en la génesis de este tipo de adenoma. De manera excepcional se ha descrito mutación en el codón 12 del gen H-ras, asociada a comportamiento agresivo (39), lo que no ha sido confirmado en series más amplias (40). En contraste, mutaciones de ras sí han sido detectadas en carcinomas hipofisarios, sugiriendo que dicha mutación podría constituir un evento en la malignización (40). En prolactinomas también se ha descrito niveles elevados de expresión de PTTG, sin que su significado esté claramente establecido (29).
ADENOMAS TIROTROPOS
Los adenomas hipofisarios secretores de hormona tirotropa (tirotropinomas) son tumores poco frecuentes que constituyen entre el 0,5 y 2% de los adenomas hipofisarios (3,41-43). Son causa infrecuente de hipertiroidismo, que puede simular enfermedad de Graves. Lo habitual es que la inmunohistoquímica demuestre, además de hormona tirotropa (TSH), la presencia de otras hormonas como alfa-subunidad. Clínicamente pueden presentar acromegalia, con secreción de GH en el 16-19% de los casos e hiperprolactinemia en un 11-21% (3,43). La hiperprolactinemia, habitualmente con cifras menores a 200 µg/ml suele ser debida a efecto de sección del tallo, especialmente en macroadenomas con extensión supraselar (44); en el 30-80% de los casos se observa elevación de la subunidad alfa de las hormonas glucoproteicas (43). La presencia de cifras elevadas de TSH permite la sospecha diagnóstica, si bien puede estar presente solo en el 41% de los casos (43). Estas neoplasias pueden presentarse a cualquier edad y al contrario que las enfermedades relacionadas con el tiroides, no tienen predilección por las mujeres.
En el momento del diagnóstico la mayoría corresponden a macroadenomas, con comportamiento localmente agresivo e invasión de estructuras paraselares (3,43), si bien en un 10% ser microadenomas (45,46).
Morfológicamente los adenomas tirotropos suelen ser tintorialmente cromófobos, con arquitectura cordonal o difusa y es frecuente que presenten fibrosis del estroma. No es inhabitual observar en estos adenomas núcleos pleomórficos y prominente nucleolo sin que ello indique malignidad (46).
Con tinciones inmunohistoquímicas se demuestra positividad para TSH (fig. 10) y en la mayoría de los casos para alfa subunidad. Es frecuente la positividad para otras hormonas adenohipofisarias y se ha documentado inmunorreactividad para GH o prolactina hasta en un 75% de los casos (44). También se ha descrito, aunque en menor proporción, positividad para hormonas gonadotropas. Por otra parte algunas series demuestran inmunorreactividad para TSH hasta en un 30% de adenomas somatotropos (47).
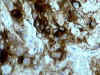
Fig. 10. Tirotropinoma. Positividad
para TSH (estreptavidina biotina peroxidasa x400).
No han sido documentadas mutaciones gsp en esta variedad de adenoma. Otros trabajos que han valorado la expresión de oncogenes como c-myc, c-fos y c-myb en adenomas hipofisarios, no encontraron sobrexpresión de los mismos en adenomas tirotropos (46). Tampoco se han identificado alteraciones en p53 (48). Un candidato que podría estar implicado en la génesis de los tirotropinomas es el factor de transcripción nuclear específico de la hipófisis Pit-1/GHF-1. En series estudiadas no encontraron mutaciones del gen POU1F1, que codifica Pit-1, en tirotropinomas pero sí sobrexpresión de la proteína, indicativo de una alteración en la regulación del gen (42,44). Otros candidatos descritos son formas mutadas para receptores de hormonas tiroideas (TR), pero los datos disponibles no son concluyentes (46). Finalmente, existen estudios que demuestran un papel de factores de crecimiento como el factor de crecimiento fibroblástico básico (FGFb). El origen tumoral del FGFb fue corroborado mediante el hallazgo de ARNm en los adenomas, sugiriéndose un posible papel autocrino de este factor en el desarrollo tumoral (49).
Tras el tratamiento quirúrgico la curación de los tirotropinomas no supera el 40% (46). También responden a radioterapia, que se indica cuando la resección ha sido incompleta. La expresión por las células neoplásicas de receptores para somatostatina justifica el tratamiento con análogos (octeótrido, lanreótido), especialmente en los casos en que la cirugía está contraindicada. Tras el tratamiento con octeótrido se ha descrito la normalización de la función tiroidea hasta en un 95% de los casos y reducción del tamaño tumoral en un 52% (43,50-52), que se acompaña en el estudio microscópico de fibrosis (45).
ADENOMAS CORTICOTROPOS
El hipercortisolismo dependiente del exceso de producción de hormona corticotropa (ACTH) por la hipófisis constituye la enfermedad de Cushing, que corresponde a dos tercios de los casos de síndrome de Cushing (3). Antes del desarrollo de las técnicas de diagnóstico por imagen muchos casos de enfermedad de Cushing eran tratados con adrenalectomía bilateral, con posterior desarrollo de un síndrome de Nelson (aumento de tamaño del adenoma hipofisario post adrenalectomía) (53). Con la radiografía de cráneo no se observa lesión en la mayoría de las ocasiones, puesto que suelen ser microadenomas. La TAC y la RMN han aumentado considerablemente la sensibilidad diagnóstica. Ante un exceso de secreción de ACTH, otros diagnósticos que deben ser tenidos en cuenta son la secreción ectópica de ACTH por otras neoplasias como el carcinoma de célula pequeña pulmonar o el tumor carcinoide (54) y la secreción ectópica de CRH (hormona liberadora de ACTH) documentada en tumores endocrinos de pulmón, páncreas, tracto gastrointestinal, tiroides, próstata y suprarrenal (55).
La mayor parte de los corticotropinomas corresponden a adenomas densamente granulados, tintorialmente basófilos y con arquitectura difusa o trabecular (1,3). Con tinciones inmunohistoquímicas presentan grados variables de positividad para ACTH, y pueden expresar otros péptidos relacionados con la POMC (proopiomelanocortina) (3,56,57) y alfa-subunidad (58). Los tumores de mayor tamaño se observan en el S. de Nelson, o corresponden a adenomas corticotropos no funcionantes (silentes) (3).
La microscopía electrónica demuestra numerosos gránulos de secreción de 150-450 nm, con contenido de densidad variable (fig. 11). Un hallazgo destacado es la presencia de numerosos filamentos de disposición perinuclear (1,59) y que corresponden a citoqueratina como puede demostrarse con inmunohistoquímica, observándose en las células del adenoma, positividad perinuclear en forma de anillo con citoqueratinas de bajo peso molecular. Otro hallazgo característico, que se puede observar en las células corticotropas no tumorales que rodean al adenoma es el cúmulo citoplasmático de filamentos de citoqueratina (60), lo que se conoce como cambio hialino de Crooke, y que se produce como consecuencia del efecto supresor de los corticoesteroides (3,61). Más infrecuentes son los adenomas pobremente granulados, de aspecto cromófobo con H&E.
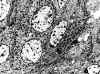
Fig. 11. Adenoma corticotropo
densamente granulado. Imagen ultraestructural (x13600).
Un 50% de los adenomas corticotropos parecen presentar alteraciones en p53, si bien no se han identificado mutaciones. Al igual que en otros tipos de AH, estas alteraciones parecen secundarias a alteraciones en la regulación de p53 nativa (62). Algunos estudios se han centrado en el papel de los genes que codifican receptores específicos de células corticotropas con actividad estimuladora como CRH-R («corticotropin releasing hormone —CRH— receptor»), receptor de vasopresina y receptor de LIF («leukaemia inhibitory factor») o inhibidora como el receptor de glucocorticoides (GR), como posibles candidatos en la génesis de los corticotropinomas; los resultados no obstante no son concluyentes (62). Otro gen relacionado es el p27 (gen supresor de tumor), cuya inactivación se ha descrito en corticotropinomas malignos. En adenomas se han observado esporádicamente bajos niveles de expresión de p27 (62).
Los carcinomas corticotropos silentes constituyen el 0,05% de los tumores hipofisarios (63). Morfológicamente pueden ser indistinguibles de los adenomas corticotropos, o bien pueden presentar características atípicas como mayor grado de pleomorfismo, actividad mitótica y mayor actividad proliferativa con Ki-67 (63).
ADENOMAS GONADOTROPOS FUNCIONANTES Y NO FUNCIONANTES
Los gonadotropinomas, sean tumores funcionantes o no, son detectados en la mayoría de los casos como consecuencia del efecto de masa intracraneal, ya que los síntomas derivados de la disfunción genital que produce el exceso de hormonas gonadotropas suelen ser poco manifiestos (64). En los pacientes de sexo masculino su diagnóstico se basa en la detección de cifras aumentadas de hormonas gonadotropas. En las mujeres premenopáusicas, estos tumores son raros y en mujeres peri o menopáusicas la diferenciación con adenomas no secretores puede ser imposible, al presentar estas pacientes elevación fisiológica de las hormonas gonadotropas. La mayoría son macroadenomas con extensión extraselar lo que determina la aparición de síntomas compresivos locales (64). La inmunohistoquímica ha permitido demostrar que la mayor parte de los adenomas descritos como «no funcionantes» corresponden a adenomas gonadotropos silentes. La inmunohistoquímica hormonal muestra en los gonadotropinomas, coexpresión de hormona luteinizante (LH), folículoestimulante (FSH) o alfa-subunidad, en porcentaje y densidad marcadamente variable (fig. 12) (65). Es frecuente observar para estas hormonas, una inmunotinción polarizada en las células de disposición perivascular (66). Las series recientes demuestran que los adenomas nulos (adenomas sin secreción ni expresión inmunohistoquímica de ninguna hormona) son infrecuentes (<5%), y que la mayoría de los adenomas no funcionantes, incluidos los oncocitomas, corresponden a adenomas gonadotropos. (3,66,67). Los adenomas gonadotropos secretores y no secretores expresan cromogranina A (68), lo que puede tener utilidad diagnóstica, especialmente en aquellos casos con negatividad inmunohistoquímica hormonal.

Fig. 12. Adenoma gonadotropo
silente. Inmunorreactividad para LH (estreptavidina biotina peroxidasa x400).
Respecto a la morfología los gonadotropinomas presentan una arquitectura altamente característica, con formación de pseudopapilas y disposición rosetoide perivascular de las células (fig. 13). Muchos de ellos presentan cambio oncocítico demostrado con microscopía electrónica (fig. 14), y que puede ser evaluado mediante inmunohistoquímica (fig. 15). En los gonadotropinomas el cambio oncocítico puede ser focal, aunque en ocasiones afecta a más del 50% de las células, siendo estos casos tipificados como oncocitomas (69).
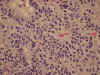
Fig. 13. Adenoma gonadotropo
silente. Arquitectura rosetoide pseudopapilar (H&E x200).

Fig. 14. Adenoma oncocítico,
imagen ultraestructural. Citoplasma ocupado por numerosas mitocondrias. Entre
las mismas se identifican gránulos de secreción (x31920).

Fig. 15. Adenoma oncocítico.
Inmunorreactividad con anticuerpo antimitocondrial (Ac 113-1, estreptavidina
biotina peroxidasa x400).
ADENOMAS NULOS
La descripción inicial de los adenomas nulos se basa en criterios de microscopía electrónica (70). De manera característica se trata de adenomas no funcionantes que no presentan signos de diferenciación en el estudio ultraestructural (fig. 16) (5). La inmunohistoquímica y las técnicas de hibridación in situ para ARN mensajero han demostrado expresión de hormonas gonadotropas en la mayoría de los casos de adenoma nulo (71,72), por lo que en la actualidad se encuadrarían dentro de los adenomas gonadotropos silentes (3).
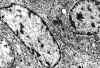
Fig. 16. Adenoma nulo. Imagen
ultraestructural que muestra orgánulos pobremente desarrollados, vesículas de
lípidos y muy escasos gránulos de secreción (x13080).
ADENOMAS NO FUNCIONANTES NO GONADOTROPOS
Los adenomas no funcionantes que expresan con inmunohistoquímica hormonas no gonadotropas constituyen un grupo heterogéneo, que incluye el adenoma somatotropo silente (73), el adenoma corticotropo silente (74), el adenoma tirotropo silente y el prolactinoma silente (75). Todos ellos son infrecuentes. Las manifestaciones clínicas son consecuencia del crecimiento local, incluyendo hiperprolactinemia derivada del efecto de sección del tallo.
Se han descrito de forma excepcional mutaciones en el gen gsp en adenomas hipofisarios no funcionantes (76). Otra mutación observada en adenomas no funcionantes afecta a Gaq, que se acompaña de la activación de la fosfolipasa C (77).
ADENOMAS PLURIHORMONALES
Una proporción significativa de AH muestra inmunorreactividad para más de una hormona (78). Las combinaciones más habituales son la asociación de GH y prolactina, o GH, prolactina con hormonas glicoproteicas, observada en adenomas somatotropos. Combinaciones menos habituales serían TSH y prolactina, y ACTH, GH con prolactina entre otras (79). La clasificación de estos casos debe basarse en la integración de datos morfológicos y la forma de presentación clínica.
ADENOMAS SIN REACTIVIDAD PARA HORMONAS HIPOFISARIAS
El estudio inmunohistoquímico hormonal permite la demostración de hormonas hipofisarias en la mayoría de los casos de AH. Sin embargo, de forma excepcional puede no ser posible demostrar inmunorreactividad para ninguna hormona. La negatividad puede ser debida a problemas técnicos derivados del procesamiento, el tipo de anticuerpo utilizado o la baja expresión de hormona. En ocasiones las técnicas de hibridación in situ demuestran ARN mensajero especialmente de hormonas gonadotropas (80). La inmunotinción con cromogranina A puede ser útil para confirmar la naturaleza endocrina del tumor (68).
MARCADORES DE AGRESIVIDAD
Los tumores hipofisarios pueden presentar crecimiento localmente agresivo, con capacidad de invadir el seno esfenoidal o el cavernoso, y de extenderse por encima de la silla turca, lo que compromete el tratamiento quirúrgico y la supervivencia libre de enfermedad. De manera excepcional pueden originar metástasis. No existen datos morfológicos convencionales que permitan establecer el comportamiento biológico de los tumores hipofisarios, si bien algunos marcadores pueden ser de utilidad. En general, los tumores con mayor actividad proliferativa medida con Ki-67 (fig. 17) presentan mayor agresividad local. Tumores hipofisarios con más del 3-4% de actividad proliferativa deberían ser considerados «atípicos» (14,81). El diagnóstico de carcinoma hipofisario solo debe ser hecho en presencia de metástasis (3,5,82). Otros marcadores de proliferación como ADN topoisomerasa II alfa han sido estudiados en tumores hipofisarios, describiéndose un aumento de expresión en adenomas silentes, en adenomas invasivos y en carcinomas (83).
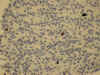
Fig. 17. Adenoma gonadotropo
silente. Actividad proliferativa con Ki-67 (estreptavidina biotina peroxidasa
x200).
Recientemente se han estudiado marcadores como NCAM («polysialylated neural cell adhesion molecule») (84), galectina-3 (85), o leptina (86) que parecen asociarse con un comportamiento más agresivo. La angiogénesis tumoral también ha sido explorada (fig. 18). Algunos estudios iniciales demostraron que los AH presentan menor grado de vascularización que la hipófisis normal (87), lo que fue confirmado posteriormente utilizando factor VIII (88) y CD31 (89) como marcadores endoteliales. No se observaron diferencias en la DMV de adenomas somatotropos, no funcionantes y macroprolactinomas. Sin embargo, los microprolactinomas parecen presentar menor DMV que los macroadenomas (89). El estudio de la expresión de factor de crecimiento endotelial vascular (VEGF), parece mostrar un mayor grado de inmunorreactividad en adenomas somatotropos, corticotropos y nulos no oncocíticos (90). Los carcinomas hipofisarios, por otra parte, presentan el mayor grado de expresión (90). Los estudios de angiogénesis en AH pueden adquirir un especial interés en relación con el efecto antiangiogénico que los análogos de somatostatina parecen presentar (91). Un aumento en la expresión de ciclooxigenasa-2 (COX-2) en los tumores hipofisarios ha sido recientemente descrito, y parece asociarse con un mayor tamaño del tumor (92). El aumento de la expresión del gen PTTG (30) y del factor de crecimiento fibroblástico 2 (FGF-2) (29) en AH también parece asociarse con un comportamiento agresivo. Recientemente se ha descrito expresión de Her-2/neu mediante inmunohistoquímica e hibridación «in situ» en dos casos de carcinoma gonadotropo (93). En resumen, la aplicación de nuevos marcadores puede aportar información que permita establecer el comportamiento de los adenomas hipofisarios, y ayudar a predecir la respuesta a determinados tratamientos.

Fig. 18. Adenoma gonadotropo
silente. Tinción de vasos con CD34 (estreptavidina biotina peroxidasa x200).
BIBLIOGRAFÍA
Kovacs K, Horvath E. Tumors of the pituitary gland. Atlas of tumor pathology, 2nd series, fascicle 21. Washington D.C.: Armed Forces Institute of Pathology, 1986.
Lloyd RV. Surgical pathology of the pituitary gland, Philadelphia,1993.
Asa SL. Tumors of the pituitary gland. Atlas of tumor pathology. Third series, fascicle 22. Washington DC: Armed Forces Institute of Pathology; 1998.
Molitch ME. Pituitary incidentalomas. Endocrinol Metab Clin North Am 1997; 26: 725-40.
Kovacs K, Scheithauer BW, Horvath E, Lloyd RV. The World Health Organization classification of adenohypophysial neoplasms. A proposed five-tier scheme. Cancer 1996; 78: 502-10.
Kovacs K, Horvath E, Vidal S. Classification of pituitary adenomas. J Neurooncol 2001; 54: 121-7.
Ezzat S, Asa SL, Stefaneanu L, Whittom R, Smyth HS, Horvath E, Kovacs K, Frohman LA. Somatotroph hyperplasia without pituitary adenoma associated with a long standing growth hormone-releasing hormone-producing bronchial carcinoid. J Clin Endocrinol Metab 1994; 78: 555-60.
Boix E, Pico A, Pinedo R, Aranda I, Kovacs K. Ectopic growth hormone-releasing hormone secretion by thymic carcinoid tumour. Clin Endocrinol (Oxf) 2002; 57: 131-4.
Aranda FI, Anaya A. Adenomas hipofisarios asociados con acromegalia. Rev Esp Patol 1991; 24: 7-16.
Horvath E, Kovacs K. Morphogenesis and significance of fibrous bodies in human pituitary adenomas. Virchows Arch B Cell Pathol 1978; 27: 69-78.
Sano T, Oshima T, Yamada S. Expression of glycoprotein hormones and intracytoplasmatic distribution of cytoqueratin in growth hormone-producing pituitary adenomas. Path Res Pract 1991; 187: 530-3.
Furuhata S, Kameya T, Otani M, Toya S. Prolactin presents in all pituitary tumors of acromegalic patients. Hum Pathol 1993; 24: 10-5.
Knosp E, Kitz K, Perneczky A. Proliferation activity in pituitary adenomas measurement by monoclonal antibody Ki-67. Neurosurgery 1989; 25: 927-30.
Jaffrain-Rea ML, Di Stefano D, Minniti G, Esposito V, Bultrini A, Ferretti E, Santoro A, Faticanti Scucchi L, Gulino A, Cantore G. A critical reappraisal of MIB-1 labelling index significance in a large series of pituitary tumours: secreting versus non-secreting adenomas. Endocr Relat Cancer 2002; 9: 103-13.
Nishioka H. Ito H, Hirano A. Immunohistochemical demonstration of oncocytes in nongonadotrophic pituitary adenomas. Virchows Arch 1999; 435: 428-33.
Vidal S, Kovacs K, Horvath E, Scheithauer BW, Kuroki T, Lloyd RV. Microvessel density in pituitary adenomas and carcinomas. Virchows Arch 2001; 438: 595-602.
Landis CA, Harsh G, Lyons J, Davis RL, McCormick F, Bourne HR. Clinical characteristics of acromegalic patients whose pituitary tumors contain mutans Gs protein. J Clin Endocrinol Metab 1990; 71: 1416-20.
Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 2001; 22: 675-705.
Adams E, Brockmeier S, Friedman E, Roth M, Buchefelder M, Falhbush R. Clinical and biochemical characteristics of acromegalic patients harboring gsp-positive and gsp-negative pituitary tumors. Neurosurgery 1993; 33: 198-2.
Barlier A, Gunz G, Zamora AJ, Morange Ramos I, Figarrella Branger D, Dufour H, Enjalbert A, Jaquet P. Pronostic and therapeutic consequences of Gs alpha mutations in somatotroph adenomas. J Clin Endocrinol Metab 1998; 83: 1604-11.
Spada A, Arosio M, Bochicchio D. Clinical , biochemical and morphological correlates in patients bearing growth hormone-secreting pituitary tumors with or without constitutively adenyl cyclase. J Clin Endocrinol Metab 1990; 71: 1421-6.
Spada A, Arosio M, Basseti M, Vallar L, Clementi E, Bazzoni N. Mutations in the alpha subunit of the stimulatory regulatory protein of adenyl cyclase (gs) in human Gh-secreting pituitary adenomas. Biochemical, clinical and morphological aspects. Pathol Res Pract 1991; 187: 567-70.
Niveiro M, Aranda FI, Boix E, Payá A, Ortega E, Alenda C, Pico A. Identification of G protein alpha subunit mutations in pituitary adenomas by single-strand conformation polymorphism (SSCP) analysis. Morphological correlation in 22 cases. Virchows Arch 2001; 439: 256.
Yang I, Park S, Ryu M, Woo J, Kim S, Kim J, Kim Y, Choi Y. Characteristics of gsp-positive growth hormone-secreting pituitary tumors in Korean acromegalic patients. Eur J Endocrinol 1996; 134: 720-6.
Asa SL, Puy LA, Lew AM, Sundmark VC, Elsholtz HP. Cell type-specific expression of the pituitary transcription activator pit-1 in the human pituitary and pituitary adenomas. J Clin Endocrinol Metab 1993; 77: 1275-80.
Sanno N, Teramoto A, Matsuno A, Osamura RY. Expression of human Pit-1 product in the human pituitary and pituitary adenomas. Immunohistochemical studies using an antibody against synthetic human Pit-1 product. Arch Pathol Lab Med 1996; 120: 73-7.
Pellegrini I, Barlier A, Gunz G, Figarella-Branger D, Enjalbert A, Grisoli F, Jaquet P. Pit-1 gene expression in the human pituitary and pituitary adenomas. J Clin Endocrinol Metab 1994; 79: 189-96.
McCabe CJ, Heaney AP. Pituitary tumour transforming gene in endocrine cancer. Clin Endocrinol (Oxf) 2003; 58: 673-82.
McCabe CJ, Khaira JS, Boelaert K, Heaney AP, Tannahill LA, Hussain S, Mitchell R, Olliff J, Sheppard MC, Franklyn JA, Gittoes NJ. Expression of pituitary tumour transforming gene (PTTG) and fibroblast growth factor-2 (FGF-2) in human pituitary adenomas: relationships to clinical tumour behaviour. Clin Endocrinol (Oxf) 2003; 58: 141-50.
Zhang X, Horwitz GA, Heaney AP, Nakashima M, Prezant TR, Bronstein MD, Melmed S. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab 1999; 84: 761-7.
Sheppard MC. Primary medical therapy for acromegaly. Clin Endocrinol (Oxf) 2003; 58: 387-99.
Lucas T, Astorga R, Catala M; Spanish Multicentre Lanreotide Study Group on Acromegaly. Preoperative lanreotide treatment for GH-secreting pituitary adenomas: effect on tumour volume and predictive factors of significant tumour shrinkage. Clin Endocrinol (Oxf) 2003; 58: 471-81.
Calle-Rodrigue RD, Giannini C, Scheithauer BW, Lloyd RV, Wollan PC, Kovacs KT, Stefaneanu L, Ebright AB, Abboud CF, Davis DH. Prolactinomas in male and female patients: a comparative clinicopathologic study. Mayo Clin Proc 1998; 73: 1046-52.
Horvath E, Kovacs K. Misplaced exocytosis. Distinct ultrastructural feature in some pituitary adenomas. Arch Pathol 1974; 97: 221-4.
Ma W, Ikeda H, Yoshimoto T. Clinicopathologic study of 123 cases of prolactin-secreting pituitary adenomas with special reference to multihormone production and clonality of the adenomas. Cancer 2002; 95: 258-66.
Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, Cougard P, Chambe B, Montvernay C, Calender A. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 2002; 87: 457-65.
Hinton DR, Polk RK, Linse KD, Weiss MH, Kovacs K, Garner JA. Characterization of spherical amyloid protein from a prolactin-producing pituitary adenoma. Acta Neuropathol (Berl) 1997; 93: 43-9.
Horvath E, Kovacs K, Singer W, Smyth HS, Killinger DW, Erzin C, Weiss MH. Acidophil stem cell adenoma of the human pituitary: clinicopathologic analysis of 15 cases. Cancer 1981; 47: 761-71.
Karga HJ, Alexander JM, Hedley-Whyte ET, Klibansky A, Jameson Jl. Ras mutations in human pituitary tumors. J Clin Endocrinol Metab 1992; 74: 914-9.
Cai WY, Alexander JM, Hedley-Whyte ET, Scheithauer BW, Jameson JL, Zervas NT, Klibanski A. Ras mutation in human prolactinomas and pituitary carcinomas. J Clin Endocrinol Metab 1994; 78: 89-93.
Girod C, Trouillas J, Claustrat B. The human thyrotropic adenoma: pathologic diagnosis in five cases and critical review of the literature. Semin Diagn Pathol 1986; 3: 58-68.
Bertholon-Gregoire M, Trouillas J, Guigard MP, Loras B, Tourniaire J. Mono- and plurihormonal thyrotropic pituitary adenomas: pathological, hormonal and clinical studies in 12 patients. Eur J Endocrinol 1999; 140: 519-27.
Socin HV, Chanson P, Delemer B, Tabarin A, Rohmer V, Mockel J, Stevenaert A, Beckers A. The changing spectrum of TSH-secreting pituitary adenomas: diagnosis and management in 43 patients. Eur J Endocrinol 2003; 148: 433-42.
Sanno N, Teramoto A, Matsuno A, Inada K, Itoh J, Osamura RY. Clinical and immunohistochemical studies on TSH-secreting pituitary adenoma: its multihormonality and expression of Pit-1. Mod Pathol 1994; 7: 893-9.
Niveiro M, Meoro A, Chulia MT, Ortega E, Pico A, Aranda FI. Microadenoma hipofisario productor de TSH. Estudio morfológico tras tratamiento con lanreotido. Rev Esp Patol 2001; 34: 59-63.
Beck-Preccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD. Thyrotropin-secreting pituitary tumors. Endocr Rev 1996; 17: 610-38.
Aylwin SJ, King A, Blenke A, Geddes JF, Wood DF, Monson JP, Burrin JM. Free alpha-subunit and intact TSH secretion in vitro are closely associated in human somatotroph adenomas. Eur J Endocrinol 1998; 139: 378-86.
Summi T, Stefaneanu L, Kovacs K, Asa SL, Rindi G. Immunohistochemical study of p53 protein in human and animal pituitary tumors. Endocrinol Pathol 1993; 4: 95-99.
Ezzat S, Horvath E, Kovacs K, Smyth HS, Singer W, Asa SL. Basic fibroblastic growth factor expression by two prolactin and tyrothropin producing pituitary adenomas. Endocrinol Pathol 1995; 6: 125-34.
Sy RA, Bernstein R, Chynn KY, Kourides IA. Reduction in size of a thyrotropin and gonadotropin secreting pituitary adenoma treated with octreotide acetate (somatostatin analog). J Clin Endocrinol Metab 1992; 74: 690-4.
Lee EJ, Kim KR, Lim SK, Lee HC, Kim DI, Kim SH, Huh KB. Reduction in size of a thyrotropin-secreting pituitary adenoma treated with octreotide acetate (somatostatin analog). Eur J Endocrinol 1994; 131: 109-12.
Iglesias P, Díez JJ. Long-term preoperative management of thyrotropin-secreting pituitary adenoma with octreotide. J Endocrinol Invest 1998; 21: 775-8.
Nelson DH, Meakin JW, Thorn GW. ACTH-producing pituitary tumors following adrenalectomy for Cushing syndrome. Ann Intern Med 1960; 52: 560-9.
Aranda FI. Metástasis suprarrenales por tumor carcinoide atípico bronquial productor de ACTH. Rev Esp Patol 1996; 29: 81-3.
Asa SL, Kovacs K, Vale W, Petrusz P, Vecsei P. Immunohistologic localization of corticotrophin-releasing hormone in human tumors. Am J Clin Pathol 1987; 87: 327-33.
Nagaya T, Seo H, Kuwayama A, Sakurai T, Tsukamoto N, Nakane T, Sugita K, Matsui N. Pro-opiomelanocortin gene expression in silent corticotroph-cell adenoma and Cushing’s disease. J Neurosurg 1990; 72: 262-7.
Stefaneanu L, Kovacs K, Horvath E, Lloyd RV. In situ hybridization study of pro-opiomelanocortin (POMC) gene expression in human pituitary corticotrophs and their adenomas. Virchows Arch A Pathol Anat Histopathol 1991; 419: 107-13.
Berg KK, Scheithauer BW, Felix I, Kovacs K, Horvath E, Klee GG, Laws ER Jr. Pituitary adenomas that produce adrenocorticotropic hormone and alpha-subunit: clinicopathological, immunohistochemical, ultrastructural, and immunoelectron microscopic studies in nine cases. Neurosurgery 1990; 26: 397-403.
Horvath E, Kovacs K, Josse R. Pituitary corticotroph cell adenoma with marked abundance of microfilaments. Ultrastruct Pathol 1983; 5: 249-55.
Neumann PE, Horoupian DS, Goldman JE, Hess MA. Cytoplasmic filaments of Crooke’s hyaline change belong to the cytokeratin class. An immunocytochemical and ultrastructural study. Am J Pathol 1984; 116: 214-22.
Felix IA, Horvath E, Kovacs K. Massive Crooke’s hyalinization in corticotroph cell adenomas of the human pituitary. A histological, immunocytological, and electron microscopic study of three cases. Acta Neurochir (Wien) 1981; 58: 235-43.
Dahia PL, Grossman AB. The molecular pathogenesis of corticotroph tumors. Endocr Rev 1999; 20: 136-55.
Roncaroli F, Scheithauer BW, Young WF, Horvath E, Kovacs K, Kros JM, Al-Sarraj S, Lloyd RV, Faustini-Fustini M. Silent corticotroph carcinoma of the adenohypophysis: a report of five cases. Am J Surg Pathol 2003; 27: 477-86.
Young WF Jr, Scheithauer BW, Kovacs KT, Horvath E, Davis DH, Randall RV. Gonadotroph adenoma of the pituitary gland: a clinicopathologic analysis of 100 cases. Mayo Clin Proc 1996; 71: 649-56.
Martinez AJ. The pathology of nonfunctional pituitary adenomas. Semin Diagn Pathol 1986; 3: 83-94.
Ho DMT, Hsu CY, Ting LT, Chiang H. The clinicopathological characteristics of gonadotroph cell adenoma: a study of 118 cases. Hum Pathol 1997; 28: 905-11.
Günzl HJ, Saeger W, Diehl S, Lüdecke DK. Immunohistochemical analyses of oncocytic and cromophobe pituitary adenomas. Exp Clin Endocrinol 1988; 92: 51-8.
Vidal N, Paules MJ, Villabona C, Gómez JM, Ferrer I. Cromogranina A en adenomas hipofisarios humanos. Rev Esp Patol 2003; 36: 189-94.
Yamada S, Asa SL, Kovacs K. Oncocytomas and null cell adenomas of the human pituitary: morphometric and in vitro functional comparison. Arch Pathol A Pathol Anat 1988; 413: 333-9.
Kovacs K, Horvath E, Ryan N, Ezrin C. Null cell adenoma of the human pituitary. Virchows Arch A Pathol Anat Histol 1980; 387: 165-74.
Asa SL, Gerrie BM, Singer W, Horvath E, Kovacs K, Smyth HS. Gonadotropin secretion in vitro by human pituitary null cell adenomas and oncocytomas. J Clin Endocrinol Metab 1986; 62: 1011-9.
Lloyd RV, Jin L, Fields K, Chandler WF, Horvath E, Stefaneanu L, Kovacs K. Analysis of pituitary hormones and chromogranin A mRNAs in null cell adenomas, oncocytomas, and gonadotroph adenomas by in situ hybridization. Am J Pathol 1991; 139: 553-64.
Kovacs K, Lloyd R, Horvath E, Asa SL, Stefaneanu L, Killinger DW, Smyth HS. Silent somatotroph adenomas of the human pituitary. A morphologic study of three cases including immunocytochemistry, electron microscopy, in vitro examination, and in situ hybridization. Am J Pathol 1989; 134: 345-53.
Horvath E, Kovacs K, Killinger DW, Smyth HS, Platts ME, Singer W. Silent corticotropic adenomas of the human pituitary gland: a histologic, immunocytologic, and ultrastructural study. Am J Pathol 1980; 98: 617-38.
Sakurai T, Seo H, Yamamoto N, Nagaya T, Nakane T, Kuwayama A, Kageyama N, Matsui N. Detection of mRNA of prolactin and ACTH in clinically nonfunctioning pituitary adenomas. J Neurosurg 1988; 69: 653-9.
Tordjman K, Stern N, Ouaknine G, Yossiphov Y, Razon N, Nordenskjöld M, Friedman E. Activating mutations of the Gs alpha-gene in nonfunctioning pituitary adenomas. J Clin Endocrinol Metab 1993; 77: 765-9.
Oyesiku NM, Evans CO, Brown MR, Blevins LS, Tindall GT, Parks JS. Pituitary adenomas: screening for G alpha q mutations. J Clin Endocrinol Metab 1997; 82: 4184-8.
Scheithauer BW, Horvath E, Kovacs K, Laws ER Jr, Randall RV, Ryan N. Plurihormonal pituitary adenomas. Semin Diagn Pathol 1986; 3: 69-82.
Ho DM, Hsu CY, Ting LT, Chiang H. Plurihormonal pituitary adenomas: immunostaining of all pituitary hormones is mandatory for correct classification. Histopathology 2001; 39: 310-9.
Sanno N, Jin L, Qian X, Osamura RY, Scheithauer BW, Kovacs K, Lloyd RV. Gonadotropin-releasing hormone and gonadotropin-releasing hormone receptor messenger ribonucleic acids expression in nontumorous and neoplastic pituitaries. J Clin Endocrinol Metab 1997; 82: 1974-82.
Thapar K, Kovacs K, Scheithauer BW, Stefaneanu L, Horvath E, Pernicone PJ, Murray D, Laws ER Jr. Proliferative activity and invasiveness among pituitary adenomas and carcinomas: an analysis using the MIB-1 antibody. Neurosurgery 1996; 38: 99-106.
Pernicone PJ, Scheithauer BW, Sebo TJ, Kovacs KT, Horvath E, Young WF Jr, Lloyd RV, Davis DH, Guthrie BL, Schoene WC. Pituitary carcinoma: a clinicopathologic study of 15 cases. Cancer 1997; 79: 804-12.
Vidal S, Kovacs K, Horvath E, Rotondo F, Kuroki T, Lloyd RV, Scheithauer BW. Topoisomerase II alpha expression in pituitary adenomas and carcinomas: relationship to tumor behavior. Mod Pathol 2002; 15: 1205-12.
Trouillas J, Daniel L, Guigard MP, Tong S, Gouvernet J, Jouanneau E, Jan M, Perrin G, Fischer G, Tabarin A, Rougon G, Figarella-Branger D. Polysialylated neural cell adhesion molecules expressed in human pituitary tumors and related to extrasellar invasion. J Neurosurg 2003; 98: 1084-93.
Riss D, Jin L, Qian X, Bayliss J, Scheithauer BW, Young WF Jr, Vidal S, Kovacs K, Raz A, Lloyd RV. Differential expression of galectin-3 in pituitary tumors. Cancer Res 2003; 63: 2251-5.
Isono M, Inoue R, Kamida T, Kobayashi H, Matsuyama J. Significance of leptin expression in invasive potential of pituitary adenomas. Clin Neurol Neurosurg 2003; 105: 111-6.
Schechter J. Ultrastructural changes in the capillary bed of human pituitary tumours. Am J Pathol 1972; 67: 109-26.
Jugenburg M, Kovacs K, Stefaneanu L, Scheithauer BW. Vasculature in nontumorous hypophyses, pituitary adenomas, and carcinomas: a quantitative morphologic study. Endocr Pathol 1995; 6: 115-24.
Turner HE, Nagy Z, Gatter KC, Esiri MM, Harris AL, Wass JA. Angiogenesis in pituitary adenomas and the normal pituitary gland. J Clin Endocrinol Metab 2000; 85: 1159-62.
Lloyd RV, Scheithauer BW, Kuroki T, Vidal S, Kovacs K, Stefaneanu L. Vascular Endothelial Growth Factor (VEGF) expression in human pituitary adenomas and carcinomas. Endocr Pathol 1999; 10: 229-35.
Garcia de la Torre N, Wass JA, Turner HE. Antiangiogenic effects of somatostatin analogues. Clin Endocrinol (Oxf) 2002; 57: 425-41.
Vidal S, Kovacs K, Bell D, Horvath E, Scheithauer BW, Lloyd RV. Cyclooxygenase-2 expression in human pituitary tumors. Cancer 2003; 97: 2814-21.
Roncaroli F, Nose V, Scheithauer BW, Kovacs K, Horvath E, Young WF Jr, Lloyd RV, Bishop MC, Hsi B, Fletcher JA. Gonadotropic pituitary carcinoma: HER-2/neu expression and gene amplification. Report of two cases. J Neurosurg 2003; 99: 402-8.
![]()