
Vol. 37, n.º 2, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 2, 2004 |
Carmen Bellas Menéndez
Hospital Universitario Puerta de Hierro. Madrid. cbellas.hpth@salud.madrid.org
RESUMEN
El linfoma Hodgkin es uno de los linfomas malignos más frecuentes en los países occidentales. Se caracteriza morfológicamente por la especial composición del infiltrado, en el que las células neoplásicas son minoritarias, siendo mayoritario el componente no neoplásico acompañante. En base a la morfología de las células neoplásicas, al inmunofenotipo y a la composición del infiltrado inflamatorio acompañante, se reconocen dos entidades biológicamente distintas, el linfoma Hodgkin clásico y el linfoma Hodgkin predominio linfocítico nodular.
Palabras clave: Linfoma Hodgkin, diagnóstico, inmunohistoquímica, células HRS, virus Epstein-Barr.
SUMMARY
Hodgkin’s lymphoma is one of the most common malignant lymphomas in the western world. The characteristic morphologic feature of LH is the cellular composition of the tumor tissue containing a small number of neoplastic Reed- Sternberg cells and a major population of nonneoplastic inflammatory background. On the basis of tumor cell morphology, immunophenotype, and the composition of the cellular background, two separate biologic entities, namely, classical HL and lymphocyte predominant HL have been recognized.
Key words: Hodgkin’s lymphoma, diagnosis, immunohistochemistry, HRS cells, Epstein-Barr virus.
INTRODUCCION
El linfoma Hodgkin (LH) se diferencia de la mayoría de las neoplasias malignas en su especial composición celular de forma que en la masa tumoral las células neoplásicas son minoritarias, estando el componente mayoritario constiudo por células inflamatorias. Las células de Hodgkin y de Reed-Sternberg (HRS) y sus variantes constituyen menos del 1% de la celularidad total y el componente no neoplásico esta constituido por linfocitos, histiocitos, eosinófilos y plasmáticas. La presencia de este componente sugiere que en esta neoplasia la reacción inmunológica específica es una parte importante de la enfermedad. El hecho de que las células linfoides atípicas que proliferan en el LH sean minoritarias, es la causa de que dichas células hayan sido muy difíciles de clasificar.
Desde el punto de vista clínico el LH se manifiesta por el aumento del tamaño de un ganglio linfático o grupo de ellos. La historia natural de la enfermedad (no tratada) lleva a la diseminación a grupos ganglionares vecinos, con afectación de hígado, bazo y médula ósea. Tanto las características clínicas como la respuesta al tratamiento de este tipo de linfoma, son diferentes a la de los procesos linfoproliferativos malignos denominados LNH.
Los distintos tipos de LH se diferencian en la morfología de las células RS, en la composición del infiltrado reactivo y en sus características epidemiológicas, clínicas y en la historia natural de la enfermedad. Hay cinco subtipos de LH que se denominan: esclerosis nodular (EN), celularidad mixta (CM), LH rico en linfocitos (LHRL), depleción linfocitaria (DL) y linfoma Hodgkin predominio linfocitico (LHPL).
La diferencia más importante entre las antiguas clasificaciones del LH y la más recientemente propuesta por la OMS (1), es que en ésta se reconocen dos formas diferentes de LH : el LH clásico y el LH predominio linfocítico nodular (LHPLN) conocido también como paragranuloma nodular. El LH clásico engloba a los tipos EN, CM, LHRL y DL. El predominio de linfocitos en el componente no neoplásico no es suficiente para clasificar un caso como LHPLN. Existen casos con morfología e inmunofenotipo de LH clásico que contienen un predominio de linfocitos T en el infiltrado acompañante. Estos casos en la actualidad son clasificados como LH clásico rico en linfocitos.
LINFOMA HODGKIN CLÁSICO
a) Características clínicas y morfológicas
El linfoma Hodgkin clásico incluye la esclerosis nodular, la celularidad mixta, la enfermedad de Hodgkin clásica rica en linfocitos y la depleción linfocitaria.
La EN es el subtipo más frecuente de LH (60-80% de los casos). Incide en adolescentes y adultos jóvenes aunque puede aparecer a cualquier edad. La afectación mediastínica y supradiafragmática son las localizaciones más frecuentes.
En la esclerosis nodular se observa un patrón parcialmente nodular debido a la presencia de bandas fibrosas junto a áreas difusas. La célula característica es la variante lacunar de la célula RS (fig. 1).
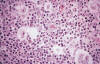
Fig. 1: Linfoma Hodgkin clásico tipo
Esclerosis Nodular. Células lacunares.
Estas células tienen un núcleo multilobulado, con nucleolos pequeños y abundante citoplasma pálido que se retrae en el tejido fijado en formol y produce un espacio vacío ¨una laguna¨. Las células lacunares suelen ser abundantes, se observan también células RS, pero éstas suelen ser escasas. El componente no neoplásico contiene linfocitos mayoritariamente de estirpe T, histiocitos, plasmáticas, eosinófilos y neutrófilos (2). Es frecuente la presencia de necrosis siendo más numerosas las células neoplásicas alrededor de los focos necróticos.
Una variante de EN es la forma sincitial de la EN. Se caracteriza porque de forma focal, se observan grandes agregados de células lacunares (3). No parece que el pronóstico de esta variante sea diferente al de la forma típica, aunque hay trabajos que apoyan que estos tipos de EN ricos en células neoplásicas y con depleción del componente no neoplásico, se asocian a masas mediastínicas grandes y estadios avanzados (4). La EN se ha graduado (grado I y grado II) basándose en el número y atipia de las células neoplásicas en los nódulos. Los grados II se superponen a la EN sincitial y a las variantes depleccionadas de linfocitos (5-7). No parece que la EN tipo II tenga relación con la supervivencia global, pero sí parece que se asocia con la supervivencia en los pacientes que recaen, lo que sugiere que estas formas se podrían beneficiar de tratamientos más agresivos (8,9).
La celularidad mixta constituye el 15-30% de los casos de LH, aparece a cualquier edad. La afectación del mediastino es poco frecuente y sin embargo la afectación del bazo y de los ganglios abdominales es más común.
En el LH celularidad mixta el infiltrado es difuso, las células neoplásicas son del tipo RS clásico (fig. 2). Estas células son bi o multinucleadas con nucléolos grandes, eosinófilos que semejan inclusiones virales. El infiltrado contiene linfocitos T, histiocitos, eosinófilos, neutrófilos y plasmáticas (2).
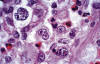
Fig. 2: Linfoma Hodgkin clásico (LHc).
Célula de Reed-Sternberg binucleada.
Tanto la EN como la CM pueden afectar focalmente a las áreas interfoliculares de los ganglios linfáticos y se pueden acompañar de hiperplasia folicular con cambios involutivos que simulan enfermedad de Castleman.
En el LH rico en linfocitos las células neoplásicas son de tipo clásico o lacunar y el componente no neoplásico está constuido mayoritariamente por linfocitos. Un porcentaje pequeño de estos casos puede tener un patrón de crecimiento vagamente nodular, con centros germinales en los nódulos y células neoplásicas en el manto de los folículos y en las áreas ínter foliculares (10). Estos casos, deben diferenciarse del LHPLN para lo cual es necesario el estudio ionmunofenotípico. Este tipo constituye aproximadamente el 6% de los casos de LH con una mayor incidencia en varones de edad media (11).
El LH tipo depleción linfocitaria es la forma menos frecuente de LH, siendo una enfermedad de ancianos y pacientes VIH seropositivos. Se presenta con linfadenopatía abdominal, hepatoesplenomegalia y afectación de la médula ósea. Tiene patrón difuso y las células neoplásicas son numerosas y de aspecto sarcomatoso siendo el infiltrado no neoplásico muy escaso (2,12).
b) Características inmunofenotípicas
Las células neoplásicas del LH clásico en la mayoría de los casos expresan CD15+, CD30+ (fig. 3), siendo negativas para CD45. La frecuencia con la que se detecta la expresión de CD15 es diferente en las distintas series probablemente debido a variaciones técnicas.
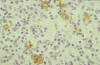
Fig. 3: Linfoma Hodgkin clásico. Expresión
del antígeno de activación CD30 en las células neoplásicas.
Solo en un pequeño número de casos se detecta en las células HRS expresión débil de diferentes antígenos de linaje B como CD20 y CD79a (13-15). Otro antígeno asociado a linaje B que se expresa en el 90% de los casos es PAX-5. Otros de los hallazgos inmunofenotípicos característicos de LH clásico es la ausencia de expresión de los factores de transcripción OCT-2 y BOB-1.
El diagnostico de LH se realiza mediante el estudio morfológico rutinario siendo los estudios inmunofenotípicos en los casos típicos no absolutamente necesario aunque forma parte de la práctica diagnóstica habitual. La ausencia de expresión conjunta de CD30 y CD15 o la expresión de CD20 intensa en las células neoplásicas obliga a reconsiderar el diagnóstico debiéndose descartar un LHPLN o un linfoma B de células grandes tipo rico en células T. La expresión genuina de antígenos de estirpe T en la HRS es absolutamente inusual. Cuando se detecta es necesario un estudio molecular del gen TCR para descartar un linfoma T.
Entre el 40-% y el 50% de los casos expresan la proteína latente de membrana (LMP-1) codificada por el VEB (fig. 4).
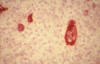
Fig. 4: Linfoma Hodgkin clásico (LHc).
Expresión fuerte de LMP-1 en las células neoplásicas.
c) Origen de las células HRS
En los estudios moleculares convencionales (Southern blot o PCR) de los genes de las inmunoglobulinas y del receptor T, en la mayoría de los casos de LH clásico no se detectan reordenamientos clonales (16-20), y solo cuando se han aplicado técnicas de micromanipulacion y se ha estudiado ADN extraído de las células neoplásicas aisladas mediante dicha técnica, se han demostrado reordenamientos monoclonales de los genes de las inmunognlobulinas (20,21). Este hallazgo prueba el origen B y el carácter maligno (clonal) de las células HRS.
Además, se han detectado mutaciones somáticas en los genes de las inmunoglobulinas reordenados (21), lo que implica que las células HRS se originan de células B centrogerminales o postcentrogerminales ya que las mutaciones se introducen en los genes de las inmunoglobulinas durante el paso de la célula B por el centro germinal.
Pero sorpresivamente las células HRS, a pesar de tener los genes de las inmunoglobulinas hipermutados, no expresan inmunoglobulina de superficie (22). Para que una célula B escape a la apoptosis en el centro germinal debe tener hipermutación somática que produzca una inmunoglobulina de superficie de alta afinidad. Pero a diferencia de las células normales del centro germinal, las células HRS no expresan inmunoglobulina de superficie y por lo tanto no pueden ser seleccionadas. Esto sugiere que sus precursoras deben ser células del centro germinal preapoptóticas que por mecanismos desconocidos han adquirido ventajas que las capacitan para escapar a la muerte por apoptosis.
Parece que hay diferentes factores implicados en el hecho de que las células HRS no expresen inmunoglobulinas de superficie; así en un grupo de casos se detectan mutaciones destructivas en los genes de las inmunoglobulinas reordenados, que producen reordenamientos no funcionales y que hacen que dichos genes no se expresen (23). También se ha reportado la falta de expresión de los factores de transcripción de los genes de las inmunoglobulinas BOB-1, OCT-2, y PU1 (24-26). La deficiencia de estos factores podría silenciar la expresión de inmunoglobulina en células HRS aunque tengan reordenamientos funcionales de dicho gen. Dichos hallazgos sugieren que las células HRS aunque derivan del centro germinal son en realidad células B defectivas debido a alteraciones en los mecanismos trasncripcionales (27).
d) Biología molecular de las células HRS
Las alteraciones moleculares por los que las células HRS escapan a la apoptosis no son bien conocidas. Las células B del centro germinal que no tiene mutaciones favorables son eliminadas mediante un mecanismo de apoptosis mediada por Fas y la células del centro germinal que expresan inmunoglobulinas de alta afinidad escapan a la apoptosis sobreexpresando el gen antiapoptótico c-FLIP (28-30). Así mientras que Fas media la apoptosis, c-Flip es un factor que protege a las células B del centro germinal frente a la misma.
Recientemente se ha descrito que las células HRS expresan constitutivamente el gen antiapoptótico c-FLIP (31). La alta concentración de c-FLIP en las células HRS podría explicar la resistencia de estas células a la apoptosis mediada por Fas. Otro mecanismo que se ha sugerido podría favorecer la resistencia a la apoptosis de la células HRS es la expresión de antígenos no linaje específicos (CD15, CD30) y la falta de expresión de antígenos específicos de linaje B, hechos ambos que favorecerían el que las HRS no fueran reconocidas en el centro germinal (27).
Estudios recientes han demostrado actividad constitutiva de NFkB en las células HRS. NFkB es un factor de trascripción nuclear que activa la expresión de genes propoliferativos y antiapoptóticos (32,33). La expresión constitutiva de NFkB dotaría a las células HRS de capacidad para crecer independientemente de las señales reguladoras, pudiendo ser éste el evento transformante principal para el desarrollo de esta neoplasia. En algunos casos de LH el mecanismo por el cual las células neoplásicas expresan constitutivamente este factor de trascripción parece estar relacionados con mutaciones en el gen Ikba (34), que codifica la molécula que en su forma salvaje (no mutada) retiene a NFkB en el citoplasma. Otra posible vía de activación constitutiva de NFkB en el LH podría estar asociada con la amplificación del locus NFkB/REL (35).
e) Patogénesis
Diferentes estudios epidemiológicos llevaron a sospechar que en el desarrollo del LH podría estar implicado algún agente infeccioso. El patrón de incidencia bimodal sugirió que la posibilidad de desarrollar la enfermedad aumentaba al retrasarse el contacto con el agente infeccioso. Este paralelismo entre la epidemiología del LH y la poliomielitis apuntaba la posibilidad de que el agente infeccioso fuera un virus. Estudios serológicos en pacientes con LH demostraron niveles elevados de anticuerpos frente al virus Epstein-Barr (VEB) especialmente anti-EBNA-2 (36). Se observó también, que el haber padecido mononucleosis infecciosa incrementaba el riesgo de desarrollar LH hasta tres veces. Estos hallazgos hicieron que las sospechas se centrasen en el VEB como el agente etiológico.
En 1987 Weiss y col. (37) detectan ADN del VEB en muestras de LH. En 1989 Weiss y col. (38) mediante técnicas de hibridación in situ (HIS) demostraron ADN del VEB en las células HRS y en 1993 Armstrong y col. (39) utilizando una técnica de HIS para la detección de EBERs demostraron que aproximadamente en el 50% de los casos de LH clásico, la mayoría de la células diagnósticas expresaban estos RNAs (fig. 5) que el VEB codifica en un gran número de copias en las células infectadas de forma latente. Trabajos posteriores demostraron que el VEB persistía en las células neoplásicas en las múltiples recaídas y en las diferentes localizaciones. En este 50% de casos las HRS expresan las proteínas codificadas por los genes LMP-1, LMP-2a y EBNA-1, patrón de expresión característico de la infección viral latente (latencia de tipo 2) (40,41).
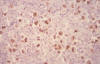
Fig. 5: Linfoma Hodgkin clásico (LHc).
Expresión de EBER en el núcleo de las células neoplásicas.
Las LMPs son proteínas virales con capacidad transformante. Este hecho soporta que el VEB en el LH no deba ser un mero pasajero, pudiendo ser un factor patogénico al menos en ese 50% de casos.
La LMP-1 simula un receptor CD40 activado constitutivamente que aumenta la actividad de la familia de los factores de trascripción NF-kB (42). La LMP-2 tiene capacidad de bloquear la expresión del receptor de la célula B y por lo tanto actúa como un gen antiapoptótico (permite que la célula no sea reconocida). Además LMP-2 también activa la familia NF-kB (43). Mediante la expresión de estas proteínas, el VEB permite que las HRS sobrevivan en el centro germinal y favorece la expresión de NFkB que consecutivamente provoca la expresión de genes proproliferativos y antiapoptóticos.
Podría ser que la imposibilidad de detectar VEB en los casos negativos, se debiera a la integración de fragmentos de genoma del virus en el genoma de la célula huésped, y que su efecto transformante en estos casos fuera del tipo impacto y carrera («hit and run») pero esto no ha podido ser demostrado (44). Por lo que hay que concluir que en al menos la mitad de los casos de LH clásico el agente primario transformante no se conoce.
Se han encontrado mutaciones del gen IkBk en un grupo de casos de LH negativos para el VEB (45). La quinasas IkB y especialmente la IkBa retienen en el citoplasma a NFkB, cuando esta quinasas reciben la señal estimuladora se fosforilizan y liberan a NFkB que se traslada al núcleo iniciando la trascripción. Las mutaciones el gen IkBa provocarían la traslocación al núcleo de NFkB en ausencia de señales estimuladoras produciendo la sobreexpresión de los genes de proliferación, de control del ciclo celular y de apoptosis que este factor de trascripción regula. Las mutaciones en el gen IkBa podrían ser un factor transformante central en los casos de LH no asociados al VEB.
Trabajos recientes han detectado amplificación de locus NFkB/REL en un grupo de casos de LH clásico (35,46) Esta alteración provoca la desregulación de NFkB y podría ser un mecanismo de activación alternativo de NFkB que actuaría en el grupo de casos de LH clásico no asociados al VEB y sin mutaciones en el gen IkBa.
LINFOMA HODGKIN PREDOMINIO LINFOCITICO NODULAR
a) Características clínicas y morfológicas
Aunque el LHPLN se parece a los otros tipos de LH en la especial composición celular, con una minoría de células neoplásicas sobre un fondo constituido por células inflamatorias benignas, difiere del LH clásico por su morfología, sus características inmunofenotípicas y por su manifestaciones clínicas (47-49).
El LHPLN en la actualidad se define por tener un patrón de crecimiento nodular que ocupa al menos el 30% del ganglio afecto con o sin áreas difusas. La variante de célula RS que lo define, se caracteriza por poseer un núcleo vesicular polilobulado con nucléolos pequeños generalmente periféricos sin halo perinucleolar. Estas células se denominan células L-H o células en «palomita de maíz» (fig. 6) (50). El fondo inflamatorio está constituido predominantemente por linfocitos acompañados de acúmulos de histiocitos mientras que las plasmáticas, los eosinofilos y neutrofilos generalmente no están presentes en el infiltrado, así como tampoco las células HRS de tipo clásico. Ocasionalmente se observa esclerosis similar a la de la EN (51).
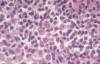
Fig. 6: Linfoma Hodgkin predominio
linfocitico nodular (LHPLN). Célula L-H acompañada de infiltrado inflamatorio
constituido por linfocitos.
El LHPLN constituye el 5% de los casos de LH. Típicamente afecta a pacientes generalmente varones, entre los 25-45 años de edad y suele afectar a ganglios periféricos respetando el mediastino. El 80% de los pacientes están en estadios iniciales en el momento del diagnóstico y el 90% de los pacientes hacen remisiones completas después del tratamiento. Las recaídas aparecen con igual frecuencia que en el LH clásico, pero las recaídas tardías y múltiples son más frecuentes que en los otros tipos de LH aunque suelen ser recaídas ganglionares aisladas que no se asocian con menor supervivencia. Los pacientes con LHPLN tienen un riesgo mayor de desarrollar LNH que los pacientes con otros tipos de LH. En las diferentes series se describe que entre un 2% y un 2,5% de pacientes con LHPLN desarrollan un LNH de células B (52-54).
b) Características inmunofenotípicas
El LHPLN se define por el inmunofenotipo. A diferencia de las células HRS del LH clásico las células L-H expresan CD45 y antígenos de estirpe B de forma que son CD20+ (fig. 7), CD79a+. Frecuentemente expresan EMA y son C15- y expresan BCL-6. La expresión débil de CD30 se observa en un porcentaje pequeño de casos (55). La expresión de OCT-2 y de BOB-1 fuerte en las células L-H es útil para diferenciar LHPLN de LH clásico.

Fig. 7: Linfoma Hodgkin predominio
linfocitico nodular (LHPLN). Las células L-H muestran expresión fuerte de CD20.
Los nódulos están constituidos mayoritariamente por linfocitos B con frecuentes células T CD57+. Estas células típicamente rodean a las células L-H conformando rosetas (56) No se detecta expresión de LMP-1.
En los nódulos se observan mallas de células dendríticas. Las áreas internodulares están predominantemente constituidas por células T.
El estudio inmunohistoquímico ayuda a reconocer el patrón de crecimiento nodular y en particular las tinciones con CD20 y CD21; esta última descubre la malla de dendríticas y muestra que los nódulos en el LHPLN son folículos o centros germinales alterados (57).
c) Origen de las células L-H
En el estudio del ADN extraído de las células neoplásicas aisladas se observan reordenamientos monoclonales de los genes de las inmunognlobulinas detectándose hipermutación somática en la región variable del gen de la IgH y también signos de mutaciones en proceso «on going». Los reordenamientos son funcionales y se detecta ARN m de Ig en las células L-H. Estos datos sugieren que las células L-H derivan de células B del centro germinal y más concretamente de centroblastos (58).
d) -Relación entre los centros germinales progresivamente transformados y el
LHPLNLa LHPLN se ha asociado persistentemente con los centros germinales progresivamente transformados (CGPT). Los CGPT se observan de forma focal en aproximadamente un 20% de casos de LHPLN (59). Los CGPT son grandes folículos que contienen numerosos linfocitos B pequeños del manto. Su semejanza con los nódulos del LHPLN ha hecho que se especule sobre la posibilidad de que el LHPLN derive de los CGPT. La mayoría de los pacientes con linfadenitis con CGPT no tienen un riesgo mayor de desarrollar LHPLN (60,61). El diagnóstico diferencial entre estas dos entidades se basa en la morfología. Los CGPT son grandes, bien delimitados, con centros germinales reactivos entre ellos mientras que los nódulos del LHPLN están pegados unos a otros, son irregulares y borran la arquitectura ganglionar. En los CGPT no se observan células L-H aunque algunos de los centroblastos que contienen pueden tener morfología similar.
BIBLIOGRAFÍA
Harris NL, Jaffe ES, Stein H et al. World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting Aerlie House Virginia , November 1997. J Clin Oncol 1999; 17: 3835-49.
Lukes R, Butler J, Hicks E. Natural history of Hodgkin’s disease as related to its pathological picture. Cancer 1966; 19: 317-44. NF-kB.
Strickler J, Michie S, Warnke R et al. The syncytial variant of nodular sclerosing Hodgkin’s disease. Am J Surg Pathol 1986; 10: 470-7.
Kant J, Hibbard S, Longo D et al. The pathological and clinical heterogeneity of lymphocyte depleted Hodgkin’s disease. J Clin Oncol 1986; 4: 284-94.
Bennet M, MacLennan K, Easterling M et al. A. The prognostic significance of cellular subtypes in nodular sclerosing Hodgkin’s disease: an analysis of 271 non-laparatomised cases (BNL1 report n.º 22). Clin Radiol 1983; 34: 497-500.
Haybittle J, Easterling B, Bennet M et al. Review of Brithish National Lymphoma Investigation studies of Hodgkin’s disease: an development of prognostic index. Lancet 1985; 1: 967-72.
MacLennan K, Bennet M, Vaughan Hudson B et al. Relationship of histopathologic features to survival and relapse in nodular sclerosing Hodgkin’s disease. Cancer 1989; 64: 1686-93.
Ferry J, Linggood R, Convery K et al. Hodgkin’s disease, nodular sclerosis type: implications of histologic subclassification. Cancer 1993; 71: 457-63.
Michel G, Bouzourene H, Delacretaz F et al. Histologic grade of nodular sclerosisng Hodgkin’s disease. Is It a prognostic factor? Kolne, Germany: 1995: 163.
Diehl V, Stein H, Sextro M et al. Lymphocyte predominat Hodgkin’s disease: a European Task Force on lymphoma projet. Blood 1996; 88: 294a.
von Vasielewski R, Mengel M, Fischer R et al. Clasical Hodgkin’s disease: clinical impact of the immunophenotype. Am J Pathol 1997; 151: 1123-30.
Neiman R, Rosen P, Lukes R. Lymphocyte depletion Hodgkin’s disease. A clinicopathological entity. N EnglJ Med 1973; 288: 751-5.
Falini B, Stein H, Pileri S et al. Expression of T cell antigens on Hodgkin’s and Reed-Sternberg cells of Hodgkin’s disease. A combined immunocytochemical and immunohistological study using monoclonal antibodies. Histopathol 1987; 12: 1129-241.
Schmid C, Pan L, Diss T et al. Expression of B cell antigens by Hodgkin’s and Reed-Sternberg cells in Hodgkin’s disease. Am J Pathol 1992; 139: 701-7.
Zukerberg L, ferry J, Harris N. Coexpression od CD15 and CD20 by Reed-Stenberg cells in Hodgkin’s disease. Am J Pathol 1991; 139: 475-83.
Weiss L Strckler J, Hu E et al. Immunoglobulin gene rearrangements in tissues involved by Hodgkin’s disease. Hum Pathol 1986; 17: 1006-14.
Sundeen J , Lipford E, Uppenkamp J et al. Rearrenged antigen receptor genes in Hodgkin’s disease. Blood 1987; 70: 96: 103-9.
Manzanal A, Santón A, Oliva H, Bellas C. Evaluation of clonal heavy chain rearrangements in Hodgkin’s disease using the polymerase chain reaction (PCR). Histopathology 1995; 27: 21-5.
Jacobsosn J, Wilkes B, Harris N. T cell receptor genes are polyclonally rearranged in Hodgkin’s disease: implications for diagnosis. Mod Pathol 1991; 4: 172-7.
Kuppers R, Rajewsky K, Zhao M et al. Hodgkin’s disease: Hodgkin’s and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to derived from B cells in at various stages of development. Proc Nat Acad Sci USA 1994; 91: 1092-6.
Kanzler H, Hansmannn ML, Kapp Uet al. Hodgkin and Reed Stenberg cells in Hodgkin’s disease represents the outgrowth of a dominant tumor clone derived from 8 crippled germinal center B cells. J Exp Med 1996; 184: 1495-505.
Marafiotti T, Hummel M, Foss HD, et al. Hodgkin and Reed Sternberg cells represent an expansion of a single clone originating from germinal center B-ceel with functional immunoglobulin gene rerrangements but defective immunoglobulin transcription. Blood 2000; 95: 1443-50.
Jox A, Zander T, Kuppers R et al. Somatic mutations within the untraslated regions of rearranged Ig genes in a case of classical Hodgkin’s disease as a potential cause for the absence of Ig in the lymphoma cells.Blood 1999; 93: 3964-72.
Re D, Muschen M, Ahmadi T et al. Oct-2 and Bob-1 deficiency in Hodgkin and Reed-Sternberg cells . Cancer Res 2001; 61: 2080-4.
Stein H, Marafioti T, Foss HD et al. Down-regulation of BoB.1/OBF.1 and Oct-2 in classical Hodgkin’s disease but not in lymphocyte predominant Hodgkin’s disease correlates with immunoglobulin transcription. Blood 2001; 97: 496-501.
Torlakovic E, Tierens A, Dang HD et al. The transcription factor PU.1, necessary for B-cell development is expressed in lymphocyte predominance, but not classical Hodgkin’s disease. Am J Pathol 2001; 159: 1807-14.
Thomas R, Re D, Wolf J, Diehl V. Part I: Hodgkin’s lymphoma-molecular biology of Reed-Sternberg cells. The Lancet Oncol 2004; 5: 11-8.
Martinez-Valdes H, Guret C, de Bouteiller O et al. Human germinal center B cells express the apoptosis-inducing genes Fas, c-myc, p53, and Bax but not the survaival gene bcl-2. J Exp Med 2001; 193: 447-58.
Hennino A, Berard M, Krammer PH et al. FLICE-inhibitory protein is a key regulator of germinal center B cell apoptosis. J Exp Med 2001; 193: 447-58.
van Eijk M, Medema J, de Groot C, et al. Cutting edge: cellular Fas –associated death domain-like –converting enzyme-inhibitory protein protects germinal center B cells from apoptosis during germinal center reactions. J Immunol 2001; 166: 6473-6.
Thomas R, Kallenborn A, Wickenhauser C et al. Constitutive expression of c-Flip in Hodgkin and Reed-Sternberg cells. Am J pathol 2002; 160: 1521-8.
Bargou RC, Emmerich F, Krappmann D et al. Constitutive nuclear factor Kappa B –Rel A activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Inves 1997; 100: 2961-9.
Hinz M, Lenke P Anagnostopoulos I et al. Nuclear factor Kappa B dependent gene expression profiling of Hodgkin’s disease tumor cells pathogenetic significance, and link to constitutive signal transducer and activator of transcripcion 5a activity. J Exp Med 2002; 196: 605-17.
Jungnickel B, Starastchek-Jox A, Brauminger A et al. Clonal deleterious mutations in the Kappa B alpha gene in the malignant cells in Hodgkin’s lymphoma. J Exp Med 2000; 191: 395-402.
Joos S, Menz CK, Wrobel G et al. Classical Hodgkin’s lymphoma is characterized by recurrent copy number gains of the short arm of chromosoma 2. Blood 2002; 99: 1381-7.
Mueller N, Evans A, Harris NL et al. Hodgkin’s disease and Epstein-Barr virus. Altered antibody pattern before diagnosis. N Engl J Med 1989; 320: 689-95.
Weiss LM, Strickler JG, Warnke RA et al. Epstein-Barr viral DNA in tissues of Hodgkin’s disease. Am J Pathol 1987; 129: 86-91.
Weis LM, Movahed RA, Warkne RA, et al. Detection of Epstein-Barr viral genomes in Reed-Sternberg cell of Hodgkin’s disease. N Eng J Med 1989; 320: 502-6.
Armstrong AA, Alexander FE, Paes RP, et al. Association of Epstein-Barr virus with pediatric Hodgkin’s disease. Am J Pathol 1993; 142: 1683-8.
Pallensen G, Hamilton Dutoit SJ, Rowe M et al. Expression of Epstein-Barr virus latent membrane gene products in tumor cells of Hodgkin’s disease. Lancet 1991; 337: 320-2.
Niedobitek G, Kremmer E, Herbst H et al. Immunohistochemical detection of the Epstein-Barr virus-encoded latent membrane protein 2A in Hodgkin’s disease and infectious mononucleosis. Blood 1997; 90: 1664-72.
Gires O, Zimber-Strobl U, Gonnella R et al. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J 1997; 16: 6131-40.
Caldwell RG, Wilson JB, Anderson SJ et al. Epstein-Barr virus LMP-2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998; 9: 405-11.
Staraschek-Jox A, Kotkowski S, Belge G et al. Detection of Epstein-Barr virus in Hodgkin-Reed-Sternberg cells: no evidence for the persistence of integrated viral fragments in Latent membrane protein 1 (LMP-1) – negative classical Hodgkin’s disease. Am J Pathol 2000; 156: 209-16.
Jarret RF, Lake A, Andrew L et al. Somatic IkBa mutations are a frequent occurrence in Hodgkin’s lymphoma. Blood 2002; 100: 4333 (abstr).
Martin-Subero JL, Gesk S, Harder L et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin’s lymphoma. Blood 2002; 99: 1474-7.
Mason D, Banks P, Chan J et al. Nodular lymphocyte predominance Hodgkin’s disease: a distinct clinico-pathological entity. Am J Surg Pathol 1994; 18: 528-30.
Coles F, Cartun R, Pastuszak W. hodgkin’s disease, lymphocyte-predominant type: immunoreactivity with B- cell antibodies. Mod pathol 1988; 1: 274-85.
Pinkus J, Said J. Hodgkin’s disease, lymphocyte predominante type, nodular further evidence for a B cell derivation: L&H variants of Reed-Stenrberg cells express L26, a pan b cell marker. Am J Pathol 1988; 133: 211-7.
Lukes RJ, Butler JJ. The pathology and nomemclature of Hodgkin’s disease.Cancer Res 1966; 26: 1063-81.
Burns B, Colby T, Dorfman R. differential diagnostic features of nodular L&H Hodgkin’s disease, including progressive transformation of germinal centres. Am J Surg Pathol 1984; 8: 253-61.
Regula D, Hoppe R, Weiss L. Nodular and diffuse types of lymphocyte predominance Hodgkin’s disease. N Engl J Med 1988; 318: 214-9.
Orlando E, Lazzarino M, Brusamolino E et al. Nodular lymphocyte predominante Hodgkin’s disease (NLPHD): Clinical behavior and pattern of progression in 66 patients. Kolne, Germany: 1995; 103.
Krayalcin G, Behm F, Geiser P et al. Lymphocyte predominant Hodgkin’s disease: clinicopathological features and results of treatment the Pediatric oncology Group experience. Med Pediatt Oncol 1997; 29: 519-25.
Poppema S. the diversity of the immunohistological staining pattern of Sternberg-Reed cells. J Histochem Cytochem 1980; 28: 788-91.
Poppema S. The nature of the lymphocytes surrounding Reed-Sternberg cells in nodular lymphocyte predominante and in other types of Hodgkin’s disease.Am J Pathol 1989; 135: 351-7.
Anagnostopoulos I, Hansmann ML, Franssila K et al. European Task Force on lymphoma project on lymphocyte predominance Hodgkin’s disese: histologic and immunohistologic analysis of submitted cases reveals 2 types of Hodgkin’s disease with nodular growth pattern and abundant lymphocytes. Blood 2000; 96: 1889-99.
Marafiotti T Hummel M, Anagnostopoulos I et al. Origen of nodular lymphocyte predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 1997; 337: 453-8.
Poppema S, Kaiserling E, Lennert K. Hodgkin’s disease with lymphocyte predominance, nodular type (nodular paragranuloma) and a progressively transformed germinal centres a cytohistological study. Histopathology 1979; 3: 295-308.
Ferry J, Zukerberg L, Harris NL. Florid progressive transformation of germinal centers in young men without progression to nodular lymphocyte predominance Hodgkin’s disease (NLPHD). Am J Surg Pathol 1992; 16: 252-8.
Hansmann M, Fellbaum C, Hui P. Progressive transformation of germinal centers with and without association to Hodgkin’s disease. Am J Clin Pathol 1990; 93: 219-26.
![]()