
Vol. 37, n.º 2, 2004
|
REVISTA
ESPAÑOLA DE
Vol. 37, n.º 2, 2004 |
Begoña Vieites Pérez-Quintela, José Manuel Suárez Peñaranda
Servicio de Anatomía Patológica. Complexo Hospitalario Universitario de Santiago de Compostela. Choupana, s/n. 15706 Santiago de Compostela.
RESUMEN
Las lesiones linfoproliferativas cutáneas, neoplásicas o reactivas, presentan con frecuencia problemas diagnósticos y de clasificación. En este artículo se repasan brevemente los aspectos más relevantes de los trastornos linfoproliferativos de células T, los más habituales en la piel, prestando particular atención a los aspectos anatomopatológicos. Además de la micosis fungoide, el linfoma más frecuente en la piel, se describen los trastornos linfoproliferativos CD30+ y, más resumidamente, otros linfomas cutáneos más raros, así como la hiperplasia linfoide de células T y las potenciales lesiones precursoras de la micosis fungoide.
Palabras clave: Linfomas cutáneos de células T, hiperplasia linfoide, clasificación.
SUMMARY
The diagnosis and precise classification of neoplastic and reactive lymphoproliferative lesions of the skin cause considerable problems for pathologists. This review summarises the most significant aspects of the skin T-cell lymphomas and pseudolymphomas. We present a short description of the histopathological characteristics of these lymphomas, particularly mycosis fungoides and large cell anaplastic CD30 positive lymphoproliferative disorders. Unusual forms of T-cell skin lymphomas are also briefly considered.
Key Words: T-cell skin lymphomas, lymphoid hyperplasia, classification.
Introducción
El diagnóstico y la clasificación de los trastornos linfoproliferativos cutáneos de células T ha sido objeto de gran debate en la literatura médica y plantea todavía problemas en un buen número de casos. El diagnóstico preciso dependerá de una adecuada correlación entre los datos clínicos, histológicos, inmunohistoquímicos y los derivados del análisis molecular de las lesiones. Esta afirmación es particularmente cierta en el caso de la micosis fungoide (MF), cuya presentación clínica y aspecto histológico pueden simular un gran número de lesiones no neoplásicas (1-3).
La incidencia global de linfomas cutáneos de células T es de alrededor de 0,36 casos/100.000 habitantes/año, lo que constituye una proporción pequeña de los linfomas no Hodgkin (4). El reconocimiento de que los linfomas que se desarrollan primariamente en la piel tienen un comportamiento más indolente que sus contrapartidas ganglionares junto con la existencia de algunas formas particulares que sólo afectan a este órgano durante una buena parte de su evolución, ha llevado al desarrollo de clasificaciones específicas para este grupo de lesiones. Los dos esquemas de clasificación empleados habitualmente son el de la Organización Mundial de la Salud (5) y el de la European Organization for Research and Treatment of Cancer (EORTC) (6), que se recogen en la tabla I. Ambas coinciden en lo esencial. Encuadran bajo el epígrafe de lesiones de comportamiento indolente a dos grandes grupos de tumores: la MF y sus variantes por un lado y, por otro, a los trastornos linfoproliferativos de células CD30+. Estos contrastan con el grupo de lesiones agresivas, constituído por el síndrome de Sèzary y el linfoma cutáneo T de células grandes CD30-. Aquí radica la principal diferencia entre ambas clasificaciones ya que tanto esta última entidad como el linfoma cutáneo T de células pleomórficas pequeñas o intermedias de la EORTC se agruparían bajo el epígrafe de linfoma periférico de células T sin especificar, de la OMS. Otras entidades más raras son incluídas en la clasificación de la EORTC como provisionales: la piel laxa granulomatosa y el linfoma de células T subcutáneo de tipo paniculítico, o todavía no son recogidas en ella, pero sí aparecen en la clasificación de la OMS: el linfoma de células T tipo citotóxico («natural killer»), el linfoma de células T gd y el linfoma epidermotrópico CD8+.

Hiperplasia linfoide de células T (Pseudolinfoma T)
El término pseudolinfoma se emplea para designar a una lesión que simula histológicamente, a veces clínicamente, un linfoma. El término carece de especificidad y debe ser empleado sólo cuándo no se pueda hacer un diagnóstico etiológico (7). Peor caracterizado y menos conocido que su contrapartida de células B, las hiperplasias linfoides de células T constituyen un grupo heterogéneo de enfermedades que deben ser separadas claramente de los linfomas cutáneos de células T. La mayoría de los casos afectan a adultos jóvenes y se trata de procesos clínicamente caracterizados por una o unas pocas lesiones del tipo placa o nódulo (7-9).
La situación que con más frecuencia presenta problemas de diagnóstico diferencial es una reacción cutánea a drogas (7), cuadro que puede ser consecuencia de la administración de un buen número de fármacos, de los cuales los anticonvulsivantes constituyen un grupo importante y con características clínico-patológicas particulares (7,10,11). El criterio clínico más importante para su diagnóstico es el hecho de que el cuadro clínico remita cuando se interrumpe la administración del fármaco. Desde un punto de vista histológico, se caracteriza por un infiltrado dérmico liquenoide o nodular, en ocasiones con células linfoides atípicas. La ausencia de epidermotropismo y mitosis, junto con la presencia de abundantes eosinófilos apuntan hacia el diagnóstico de hiperplasia linfoide, aunque la diferenciación puede resultar difícil en muchas ocasiones (3,8-11).
A veces, las dermatitis alérgicas de contacto pueden originar problemas de diagnóstico diferencial, pero la espongiosis y eosinofilia deben ser los suficientemente notorias como para evitar su confusión con la MF (3,10).
Por último, casos graves de reticuloide actínico (dermatitis actínica crónica) pueden mostrar un denso infiltrado linfoide con grupos de linfocitos intraepidérmicos que simulan microabscesos de Pautrier. La presencia de células plasmáticas y eosinófilos pueden ser una clave diagnóstica importante (10).
Lesiones precursoras de la micosis fungoide
La relación de la micosis fungoide (MF) con diversas entidades que, al menos en algún momento, se han considerado como potenciales precursores es confusa, particularmente en el caso de la denominada parapsoriasis en placas. Actualmente, se reconocen dos variedades de ésta: la parapsoriasis en placas pequeñas y la parapsoriasis en placas grandes (12).
La parapsoriasis en placas pequeñas (dermatitis crónica superficial, dermatitis digitata) afecta a individuos de mediana edad y se caracteriza por el desarrollo de lesiones eritematosas de menos de 5 cm. de diámetro (12). Histológicamente, muestra un infiltrado linfoide perivascular con ligera hiperplasia epidérmica en la que hay acantosis e hiperqueratosis con pequeños focos de paraqueratosis. En ocasiones pueden verse algunas áreas de espongiosis (12,13). A pesar de la opinión de algunos autores (14), parece claro que se trata de una lesión inflamatoria que no evoluciona hacia MF.
Por el contrario, hay datos que apoyan la opinión de que la parapsoriasis en placas grandes corresponde a una lesión con capacidad de transformación en un linfoma. Clínicamente, se caracteriza por lesiones grandes con una notable variabilidad morfológica, pero no son infrecuentes las áreas de aspecto poiquilodérmico (3). El estudio histológico muestra, en ocasiones, hallazgos indiferenciables de la parapsoriasis en placas pequeñas. Sin embargo, en las lesiones plenamente desarrolladas hay atrofia epidérmica y cierto grado de epidermotropismo (3). Se estima que entre el 10 y 30% de los casos evolucionarán hacia una MF franca (11).
Micosis fungoide
Es una neoplasia de células T maduras, de tipo colaborador, cuyas manifestaciones son preferente o exclusivamente cutáneas. Su incidencia es de 0,29 casos/100.000 habitantes/año y su etiopatogenia se desconoce (5,15).
Es la forma más común de linfoma cutáneo de células T y, a la vez, el que presenta mayores dificultades diagnósticas, particularmente la fase inicial de su desarrollo. Como ya hemos comentado la MF puede remedar una amplia variedad de procesos inflamatorios y cualquiera de los criterios diagnósticos empleados para su reconocimiento histológico puede aparecer aisladamente en otras entidades, la mayor parte de ellas procesos no neoplásicos (1-3,15-19). El problema se complica todavía más cuando consideramos el hecho, ya comentado, de que no es bien conocida la importancia que pueden tener lesiones potencialmente precursores de ella, como la parapsoriasis en placas.
El inicio de la MF tiene lugar, casi siempre, durante la vida adulta, pero pueden presentarse casos durante la adolescencia e, incluso, en la infancia. Es una forma de linfoma cuya evolución clínica sigue habitualmente el desarrollo progresivo de tres fases: parche, placa y tumoral, que refleja el inicio de la enfermedad en la epidermis y la dermis superficial (fases iniciales) para progresar con afectación en profundidad de la dermis reticular y, eventualmente, del tejido celular subcutáneo (fase tumoral) (1,16). La plétora de términos clínicos descriptivos para referirse a ella tampoco ayuda a simplificar las cosas: parapsoriasis en placas, poiquilodermia atrófica vascular, poiquilodermia prereticulocítica o parapsoriasis variegata, entre otros.
Las lesiones iniciales se describen como máculas rojizas ligeramente descamativas que se localizan en la mitad inferior del tronco, los glúteos, la parte proximal de los muslos, la cara interna de los brazos, la región periaxilar y el área submamaria. Al menos alguna de las lesiones alcanza un tamaño notable y no es raro que sobrepasen los 10 cm (16,20). En ocasiones muestra un curso intermitente, con lesiones que aparecen y desaparecen, lo que dificulta su diferenciación de un eccema (21). Una proporción desconocida de pacientes evolucionará hacia etapas más avanzadas de la enfermedad, caracterizadas por el desarrollo de placas induradas de coloración variable (desde rosa hasta parduscas), bien delimitadas y, en último término, por el desarrollo de lesiones tumorales indiferenciables de otras formas de linfomas cutáneos, que suelen indicar un estado avanzado de la enfermedad y se asocian a mal pronóstico (15,16,20,21).
En ocasiones, la enfermedad comienza con el desarrollo de este último tipo de lesiones sin pasar por la etapas previas de parche y placa. Es conocida clásicamente como la forma «d´emblée» de MF, aunque recientemente se ha planteado que podría corresponder en realidad a un linfoma pleomórfico de células T y no a una variante de MF (5,21).
En cualquier momento de la evolución de la enfermedad se puede presentar un cuadro de eritrodermia, casi siempre asociado a la presencia de adenopatías y un gran número de células neoplásicas circulantes (> 1000/mm3), conocido como síndrome de Sèzary, que complica alrededor del 3% de los casos (22-24). Una vez existe afectación ganglionar puede afectarse cualquier órgano pero, característicamente, la extensión a la médula ósea suele ser un evento tardío (21).
Sobre este patrón básico de presentación desde el punto de vista clínico existen múltiples variaciones.
El pronóstico viene condicionado fundamentalmente por el estadio evolutivo de la enfermedad, de manera que el 90% de los pacientes que muestran afectación exclusivamente cutánea del tipo parche en menos del 10% de la superficie corporal en el momento del diagnóstico sobreviven más de 15 años (4). Como ya hemos indicado, y sin que las causas sean conocidas, un grupo de pacientes permanece en la fase inicial de la enfermedad durante un largo periodo y el pronóstico en estos casos es muy bueno. Diversos factores se han asociado a mal pronóstico: ausencia de CD7 en las células neoplásicas, incremento de LDH, gran tamaño de las células neoplásicas y elevado número de células circulantes. Sin embargo, su utilidad es muy relativa ya que todos ellos suelen presentarse en fases avanzadas de la enfermedad (24). El pronóstico para los paciente con lesiones extracutáneas es malo, con una supervivencia a 5 años del 40% para pacientes con afectación ganglionar y limitada a 24-36 meses en el caso de que exista extensión visceral (4).
Características histológicas
Fase de parche: en este momento las claves diagnósticas se basan más en la arquitectura de la lesión que en las características citológicas, ya que la atipia de las células linfoides suele ser mínima y difícil de apreciar en las secciones rutinarias (10,13,21). El aspecto histológico habitual (fig. 1) es el de un infiltrado linfoide que no muestra una densidad celular particularmente alta, que afecta a la epidermis y la dermis papilar.
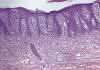
Fig.1: Micosis fungoide: imagen
característica de una fase inicial con linfocitos dispuestos a lo largo de la
unión dermo-epidérmica.
La primera suele mostrar hiperplasia psoriasiforme moderada, mientras que en la dermis hay un grado de fibrosis que debe llamar nuestra atención. Los linfocitos se disponen a lo largo de la unión dermo-epidérmica de un modo análogo a una dermatitis de interfase, aunque el cambio vacuolar es prácticamente inexistente. La extensión de células linfoides a la epidermis no se acompaña de espongiosis importante. A pesar de que la atipia citológica no suele ser un dato llamativo, los núcleos de los linfocitos en la epidermis suelen tener mayor tamaño y ser más hipercromáticos que los de los localizados en la dermis (2,10,13,17).
Fase de placa: conserva las características arquitecturales de la etapa anterior, pero se acentúan de tal manera que debe permitir el diagnóstico de la lesión por criterios estrictamente histológicos. Hay infiltrado linfoide dérmico denso, dispuesto a modo de banda a lo largo de la epidermis, la cual muestra hiperplasia psoriasiforme. El epidermotropismo de las células linfoides es la característica más destacada y los acúmulos de linfocitos intraepidérmicos (microabscesos de Pautrier) son una característica habitual en este momento (fig. 2).
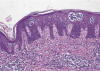
Fig.2: Micosis fungoide, fase de placa:
marcado epidermotropismo de las células linfoides con los característicos
acúmulos intraepidérmicos (microabscesos de Pautrier).
La atipia de las células linfoides puede apreciarse con facilidad y suelen verse diversas proporciones de células con aspecto inmunoblástico, células plasmáticas y eosinófilos, entremezclados con las células linfoides (2, 10,13,17).
Fase tumoral: en este momento el epidermotropismo ya no es una característica llamativa del proceso y se aprecia un denso infiltrado que ocupa la dermis papilar y reticular, que puede desplazar o destruir los anejos cutáneos. Las células linfoides son de tamaño pequeño o intermedio, con núcleos obviamente irregulares. Las células de aspecto blástico no deben superar el 25% del infiltrado, en caso contrario habría que descartar la transformación en un linfoma de alto grado (10,13,24).
La biopsia cutánea en la fase eritrodérmica es particularmente difícil de interpretar ya que el epidermotropismo es relativamente escaso (21,22).
Existen múltiples variedades histológicas de micosis fungoide, más o menos bien reconocidas: granulomatosa, folicular, siringotrópica-foliculotrópica, espongiótica, vesículo-bullosa, hipopigmentada, pustular, ulcerativa, papular-liquenoide, verrucosa, asociada a vasculitis y de tipo acantosis nigricans (3). Como puede deducirse de la gran cantidad de variedades, el aspecto histológico puede apartarse significativamente de la descripción típica que hemos realizado con anterioridad. A continuación revisaremos algunos aspectos de las más relevantes.
La forma granulomatosa se refiere a una variedad histológica caracterizada por la gran abundancia de histiocitos o macrófagos que acompaña al infiltrado linfoide, de forma intersticial o nodular, pero con características clínicas indistinguibles de la MF convencional (3,25). Puede ser difícil de diferenciar de la piel laxa granulomatosa, de hecho se han publicado casos que muestran cierta superposición clinico-patológica entre ambas entidades (26) y, en ocasiones, de dermatitis granulomatosas no neoplásicas, al menos desde el punto de vista histopatológico (25).
Es bien conocido que la afectación anexial de la MF puede producir el cuadro conocido como mucinosis folicular. Actualmente, se tiende a considerar éste como un patrón de reacción tisular y no hay características histológicas bien definidas que permitan diferenciar taxativamente entre una forma primaria y otra asociada a linfoma. El diagnóstico histológico en estos casos debe hacerse valorando la piel interfolicular (10), aunque hay formas exclusivamente foliculocéntricas, en cuyo caso se ha postulado que la ausencia de una cantidad significativa de mucina intrafolicular sugiere el diagnóstico de la variedad folicular de MF (27).
Por último, hay que señalar que la MF puede presentarse en la infancia. En esta etapa de la vida la principal diferencia clínica con la MF de adultos radica en la mayor frecuencia de lesiones hipopigmentadas. Histológicamente, se ha descrito un incremento en el número de células de Langerhans intraepidérmicas, sin que se conozca su importancia. El pronóstico no parece diferir de la forma convencional en adultos (3,28). Debe prestarse particular atención al diagnóstico diferencial con el linfoma CD30+, algo más habitual en este momento de la vida.
Inmunohistoquímica y biología molecular
El perfil inmunohistoquímico habitual de las células de la MF es el siguiente: CD2+, CD3+, CD4+, CD5+, CD7-, CD8- y CD45R0+ (5,29). Rara vez pueden presentarse casos CD4- y CD8+ (24) o CD45RA+ (29). Se ha intentado agrupar como una variedad con características propias a aquellos casos con CD8+ y CD45RA+, que podrían mostrar una presentación clínica inhabitual (erupción generalizada de placas y nódulos) y peor pronóstico, aunque es algo no aceptado en la actualidad.
El estudio del reordenamiento para el receptor de células T (TCR) se ha postulado como una herramienta diagnóstica de utilidad en la MF, vistas las dificultades que presenta el diagnóstico histológico al inicio de la enfermedad. En los últimos años se han publicado diversos estudios sobre el tema y todavía se debate su valor diagnóstico real (24,30,32). La incidencia de reordenamientos monoclonales ha oscilado entre el 50 y el 90% de las biopsias. Hay que señalar diversos aspectos que complican la interpretación de los resultados, como son que se han reseñado reordenamientos clonales en procesos no neoplásicos (papulosis linfomatoide, enfermedad de Mucha-Haberman o psoriasis), que muchos estudios se han llevado a cabo sobre material incluido en parafina y que la mayor parte de ellos emplean el estudio de TCR gamma, cuando el receptor expresado con más frecuencia por las células linfoides tanto neoplásicas como reactivas es el alfa o beta (32). Además, debe recordarse que la expansión clonal de un grupo celular no implica necesariamente el desarrollo de una neoplasia.
Síndromes linfoproliferativos de células T CD30+ primarios de la piel
Bajo este término se agrupa una serie heterogénea de entidades con diferentes características clínico-patológicas que tienen en común la infiltración cutánea por linfocitos atípicos CD30+. Dichas entidades son:
1. Linfoma anaplásico de células grandes CD30+
2. Papulosis linfomatoide
El CD30(Ki-1/Ber-H2) es un receptor transmembrana de la familia del receptor del factor de necrosis tumoral que en el tejido ganglionar normal marca las células linfoides del área parafolicular y el borde del centro folicular. Aunque originariamente el CD30 fue descrito como un marcador específico de las células de Reed-Sternberg de la enfermedad de Hodgkin, posteriormente se comprobó que marca también células B y linfocitos T activados. Algunas formas de MF y de linfomas T pleomórficos, así como ciertos infiltrados cutáneos no neoplásicos pueden ser CD30+, de ahí que dicha positividad deba valorarse con cautela (10).
Linfoma anaplásico de células grandes CD30+ primario cutáneo
El término linfoma anaplásico de células grandes (LACG) CD30+ se aplica a una forma de linfoma de células T de presentación cutánea, compuesto por células linfoides anaplásicas que se tiñen mayoritariamente con anticuerpos dirigidos contra el CD30 (5).
El LACG-CD30+, comprende aproximadamente el 25% de todos los linfomas cutáneos de células T. Puede presentarse a cualquier edad, aunque predomina en adultos y ancianos. La lesión típica consiste en nódulos o tumores solitarios, a menudo ulcerados, localizados en las extremidades. Los pacientes no suelen desarrollar síntomas sistémicos asociados, al contrario de lo que ocurre en el LACG ganglionar. En ocasiones, puede existir afectación cutánea multicéntrica, así como diseminación extracutánea, generalmente a ganglios linfáticos regionales. Las lesiones pueden regresar espontáneamente, aunque con una alta tasa de recidiva. Este es el linfoma cutáneo más común en pacientes infectados por el VIH, en los que el pronóstico es infausto (33).
Por el contrario, en la mayoría de los pacientes el pronóstico es favorable, con una supervivencia media a los 5 años de aproximadamente el 90%. Se considera factor de buen pronóstico la regresión espontánea de las lesiones y de mal pronóstico la extensión extracutánea del tumor (3,5,34,35).
Histológicamente se observa un infiltrado difuso que afecta a la dermis y al tejido celular subcutáneo, cuya extensión en profundidad puede alcanzar incluso tejidos más profundos. Está compuesto en su mayoría por células linfoides de gran tamaño (figs. 3 y 4), con abundante citoplasma basofílico, núcleo vesicular pleomórfico, con cromatina agrupada a lo largo de la membrana nuclear y nucleolo eosinófilo prominente.
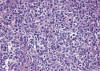
Fig. 3: Linfoma anaplásico de células
grandes (LACG) CD30+: infiltración dérmica difusa por linfocitos atípicos de
moderado-gran tamaño.

Fig. 4: Linfoma anaplásico de células
grandes (LACG) CD30+: tinción difusa de las células tumorales con CD30 (Ber-H2 /
Ki-1).
En la periferia se pueden ver linfocitos reactivos y otras células inflamatorias. Es frecuente encontrar formas atípicas, células gigantes multinucleadas y células tipo Reed-Sternberg, así como numerosas mitosis atípicas y áreas de necrosis (3,13,36). En las lesiones en regresión se observa hiperplasia epidérmica con epidermotropismo, edema dérmico y proliferación vascular (13).
Las células tumorales expresan antígenos de células T, como el CD4 y raramente CD2, CD3 y CD5. El CD30 (Ber-H2 / Ki-1) se expresa en la mayoría ((75%) de las células (3,35,37). Proteínas asociadas a gránulos citotóxicos como la granzima B, distintas perforinas y el TIA-1 se identifican en un porcentaje elevado de estos tumores (37,38). El HECA-452, que reconoce el llamado antígeno linfoide cutáneo, también muestra positividad en, aproximadamente, la mitad de las lesiones. En contra de lo que ocurre en la forma sistémica, el antígeno de membrana epitelial (EMA) es casi siempre negativo en las lesiones cutáneas primarias. La tinción con ALK (kinasa del linfoma anaplásico) es habitualmente negativa, de forma que un infiltrado ALK+ indicaría la afectación cutánea por un LACG sistémico (5,13,34,39). El CD95(APO-1/Fas) se expresa fuertemente y parece jugar un papel importante en la regresión de las lesiones (13).
En la mayoría de los casos es posible demostrar reordenamiento monoclonal de los genes del receptor de células T (TCR). La translocación t(2;5) (p23;q35) se observa en muchos casos de LACG sistémico; dicha translocación da lugar a la proteína de fusión NPM/ALK que interviene en la génesis de estas neoplasias. Sin embargo también se ha detectado en un pequeño porcentaje de los LACG primarios cutáneos, de ahí que deba ser valorada en conjunto con el resto de los hallazgos histológicos e inmunohistoquímicos (3,36,39).
El diagnóstico diferencial debe hacerse en primer lugar con las formas de LACG sistémico ya que el pronóstico de ambas entidades es sumamente distinto. En la expansión cutánea del LACG sistémico las células neoplásicas se disponen formando infiltrados dérmicos perianexiales y perivasculares (13). En ocasiones los LACG-CD30+ cutáneos primarios presentan un leve componente inflamatorio de fondo, pero si éste es muy llamativo debemos descartar la papulosis linfomatoide. Cuando los neutrófilos son predominantes, debe considerarse también la posibilidad de ciertas dermatosis pustulares o infecciosas como el pioderma gangrenoso o algunas paniculitis. Existe una variante pleomórfica de células pequeñas que puede simular el estadío tumoral de la micosis fungoide (3,5).
Algunos autores han especulado la posibilidad de que el LACG puede ser consecuencia de la progresión de otras formas de trastornos linfoproliferativos, mayoritariamente MF y también linfomas no Hodgkin T periféricos y enfermedad de Hodgkin (13,34).
Papulosis linfomatoide
La papulosis linfomatoide (PL) es una enfermedad cutánea de curso crónico y recurrente caracterizada por la aparición de pápulas y/o nódulos que histológicamente muestran un infiltrado de linfocitos T atípicos. Aunque en la mayoría de los casos sigue un curso benigno, en ocasiones puede progresar a linfoma o incluso coexistir ambos tipos de lesiones. La correlación clínico-patológica es fundamental para su correcto diagnóstico (3,40).
La afectación cutánea puede presentar forma de pápulas, nódulos, o incluso grandes placas en diferentes estadíos evolutivos. Estas lesiones suelen remitir espontáneamente en 3-6 semanas, con necrosis y ulceración que puede dar lugar a cicatrices atróficas. El número es muy variable en cada brote y, aunque lo más frecuente es que se presenten en el tronco y porción proximal de las extremidades, pueden aparecer también en la cara, cuero cabelludo, palmas y plantas. Habitualmente, afecta a mujeres en la tercera o cuarta décadas de la vida, pero se han descrito también casos en niños (13,40).
El estudio histológico de la PL demuestra un denso infiltrado dérmico compuesto por células T atípicas y una celularidad acompañante variable, con neutrófilos, eosinófilos, macrófagos y linfocitos pequeños. En función de la morfología de las células linfoides se diferencian varios tipos. La PL tipo A presenta un infiltrado dérmico difuso compuesto por linfocitos grandes, CD30+, células tipo Reed-Sternberg, y abundantes células inflamatorias (fig. 5).
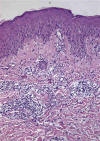
Fig. 5: Papulosis linfomatoide: imagen
característica del infiltrado dérmico compuesto por linfocitos T atípicos y las
células inflamatorias acompañantes.
En la PL tipo B el infiltrado dérmico es perivascular o «en banda» y está compuesto por linfocitos atípicos, de mediano tamaño, con núcleo cerebriforme, similares a las células de la micosis fungoide, que se acompañan de escasa inflamación y a menudo muestra epidermotropismo (3,5,13). Existe además un tercer grupo, la PL tipo C, cuyo infiltrado se compone de células linfoides de gran tamaño, atípicas, CD30+, que recuerdan las células del linfoma anaplásico de células grandes primario cutáneo y que se disponen formando grandes acúmulos, junto con escasas células inflamatorias. En este último tipo, mientras las lesiones clínicas son las típicas de la papulosis linfomatoide, la histología es característica de linfoma (3,5). La epidermis suprayacente puede mostrar espongiosis focal, ulceración, paraqueratosis o una hiperplasia pseudoepiteliomatosa (13).
Los linfocitos atípicos de la PL son CD4+ y CD8-, y a menudo expresan fenotipos aberrantes con pérdida de antígenos comunes a las células T como el CD2 y CD5 (41). Las lesiones tipo A y C expresan CD30, al contrario de las tipo B. También es habitual la expresión de ciertas proteínas asociadas a gránulos citotóxicos, mientras que la proteína ALK suele ser negativa (38,39 ).
La relación existente entre esta entidad y los linfomas se basa en el reordenamiento monoclonal del TCR que se demuestra en aproximadamente el 60% de las PL; esta clonalidad se detecta mayoritariamente en lesiones del tipo B y en algunas de las del tipo A (42). No existe hasta el momento ningún criterio capaz de predecir la capacidad de progresión de la PL a linfomas francos (MF, LACG o enfermedad de Hodgkin), a pesar de que ésto ocurre en aproximadamente el 5-10% de las PL (3,5,6,13).
Otros trastornos linfoproliferativos de
células TDentro del grupo de las enfermedades linfoproliferativas de células T existe una serie de lesiones poco frecuentes con características clínicas, histológicas e inmunofenotípicas propias, entre las que incluyen: piel laxa granulomatosa (PLG), linfoma (paniculítico) subcutáneo de células T, linfoma cutáneo ((, linfoma cutáneo CD-8 positivo y el linfoma de células NK. Estas entidades han sido y son objeto de estudio y revisión constante y su clasificación es de gran dificultad debido a su extrema rareza y a que a menudo sus características se superponen (3).
Piel laxa granulomatosa
Esta entidad muy poco frecuente constituye una forma peculiar de linfoma de células T epidermotrópico, que la OMS clasifica como una variante de la MF (5).
Clínicamente se caracteriza por una piel marcadamente engrosada en forma de pliegues flácidos y colgantes; estas lesiones se desarrollan sobre placas o pápulas eritematosas preexistentes. La localización más habitual es en las axilas y las ingles, pero también se han descrito casos con lesiones en el tronco y las extremidades (3,10,13). La enfermedad es más común en varones y suele seguir un curso indolente, aunque en ocasiones puede asociarse a linfomas de peor pronóstico como la enfermedad de Hodgkin o la MF (43).
Histológicamente, las lesiones iniciales de la PLG muestran infiltrado linfocítico perivascular en la dermis superficial y/o profunda, con ocasional epidermotropismo restringido a las capas más bajas de la epidermis, acompañado de focos de espongiosis y de hiperplasia epidérmica de aspecto psoriasiforme. Las lesiones evolucionadas se caracterizan por un infiltrado granulomatoso en el que predominan los linfocitos atípicos, hendidos, de pequeño tamaño, que se extiende por toda la dermis y alcanza el tejido celular subcutáneo. Existe, además, una segunda población compuesta por células gigantes multinucleadas y macrófagos. Las células gigantes suelen mostrar numerosos núcleos, así como fagocitosis de fibras elásticas y linfocitos. Existe edema o fibrosis en las áreas dérmicas no infiltradas y suelen verse granulomas (10,13,43).
El inmunofenotipo de las células linfoides neoplásicas se caracteriza por la positividad para CD3, CD4, CD43 y CD45RO. Las células gigantes multinucleadas expresan marcadores histiocitarios (CD68 y Mac 387). Se ha demostrado reordenamiento monoclonal del gen de la cadena ( del TCR (10,13,26,43).
El diagnóstico diferencial clínico incluye dos entidades cuyas características histopatológicas permiten una fácil diferenciación: «cutis laxa» y anetoderma. La dificultad mayor es la de distinguir la PLG de la forma granulomatosa de la MF, siempre y cuando las consideremos dos entidades diferentes. Además de la morfología de las lesiones clínicas, algunas características histológicas de esta forma de MF que permitirían diferenciarla de la PLG son el menor número de núcleos en la células gigantes, mayor atipia linfocitaria y el patrón intersticial frente al infiltrado perivascular o difuso de la PLG (10,43).
Linfoma subcutáneo de células T (paniculítico)
El linfoma subcutáneo de células T, tipo paniculítico, es una forma de linfoma de células T citotóxicas que afecta preferentemente al tejido celular subcutáneo (5). Comprende menos del 1% de los linfomas no Hodgkin y puede presentarse en un rango de edad muy amplio, con ligero predominio del sexo femenino en algunas series (5,44).
La clínica habitual consiste en nódulos subcutáneos en el tronco y las extremidades, cuyo tamaño oscila entre 0’5 y varios centímetros de diámetro. La presencia de síntomas generales es variable, pero es particularmente importante la posibilidad de desarrollar un síndrome hemofagocítico, responsable de la muerte en una buena parte de los pacientes (5,44).
El estudio histológico revela afectación difusa del tejido celular subcutáneo por un infiltrado linfoide moderadamente atípico, con extensa necrosis y en el que pueden estar presentes cariorrexis y hemofagocitosis (fig. 6).
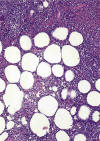
Fig. 6: Linfoma subcutáneo de células T
(paniculítico): infiltración difusa del tejido celular subcutáneo con la
disposición típica de las células neoplásicas y los focos de necrosis y
cariorrexis.
Otras características del infiltrado útiles para el diagnóstico son la disposición de las células individualmente alrededor de los adipocitos y la permeación de la pared de los vasos por células neoplásicas (44,45).
El estudio inmunohistoquímico demuestra un perfil de células T citotóxicas (positividad para CD2, CD3, CD45 y CD8) con expresión de TIA-1 o perforina. En ocasiones se encuentran variantes CD4+ o, incluso CD4-/CD8-. Los marcadores de activación de células NK (CD56 y CD57) son negativos (5,45).
El diagnóstico diferencial incluye el linfoma de células T-NK, para lo cual la inmunohistoquímica (suele mostrar postividad para CD56) y la hibridación in situ resultan muy útiles o enfermedades no neoplásicas, sobre todo la paniculitis lúpica. Por último, último es importante señalar que el antiguo término de paniculitis histiocítica citofágica no debe emplearse más que como un patrón de reacción morfológico que puede verse en trastornos linfoproliferativos y en otras situaciones que indican alteraciones inmunológicas, como el trasplante de médula ósea, infecciones, reacciones de hipersensibilidad o enfermedades del tejido conectivo (3).
Linfoma cutáneo
gdEs una neoplasia de células linfoides T que expresan un gen de la región variable 2 del receptor de células T-d (TCR d). Estas células tienen propiedades citotóxicas y suelen ser CD4 y CD8 negativas, aunque su verdadera función se desconoce (5).
La enfermedad es extremadamente rara y se caracteriza por la presencia de placas eritematosas, nódulos o tumores en la piel de tronco y extremidades. Se trata de un tumor de alto grado con un pronóstico infausto.
Histológicamente, puede presentar características similares a la MF, aunque los linfocitos tumorales no muestran el típico núcleo cerebriforme. Las áreas de necrosis y la apoptosis suelen ser llamativas.
El inmunofenotipo de las células neoplásicas es el siguiente: CD3+, CD4-, CD8-, TIA-1+, granzima-B+, perforina+, TCR d1+, CD56- y CD57-. Se ha demostrado monoclonalidad en el reordenamiento de los genes del TCR gd (3,5,46).
Linfoma cutáneo primario CD8+
Al contrario de lo que ocurre en la mayoría de los linfomas cutáneos de céulas T, en esta variedad los linfocitos son citotóxicos y expresan el marcador CD8.
La presentación clínica es muy variable y puede simular otras formas de linfomas cutáneos. Sin embargo, éste es un proceso de gran agresividad en el que existe una rápida progresión con metástasis viscerales, sin afectación ganglionar y con mortalidad elevada (3,5).
Morfológicamente, se caracteriza por un infiltrado linfoide atípico con gran epidermotropismo que puede afectar también el epitelio folicular y glandular. Los linfocitos son de mediano o gran tamaño y expresan los siguientes marcadores: CD8, CD3, CD7, TIA-1 y bF1. Son negativos para CD4, CD16, CD56 y CD57 (3,5).
Linfoma de células T-NK extranodal, tipo nasal
El linfoma de células NK, tipo nasal de presentación cutánea constituye un tumor muy infrecuente, de gran agresividad, con una mortalidad de al menos el 60% de los casos.
Las lesiones cutáneas varían desde nódulos eritematosos a tumores, úlceras, ampollas, o lesiones similares a vasculitis o paniculitis. La afectación visceral, medular o ganglionar en el momento inicial es indicador de mal pronóstico (3,5,13).
Histológicamente, el tumor consiste en un infiltrado dérmico que se extiende al tejido celular subcutáneo y que, típicamente, adquiere disposición angiocéntrica, a menudo con destrucción de los vasos afectados, de ahí el término «linfoma angiocéntrico» utilizado con anterioridad por la clasificación REAL de los linfomas (47). Las células neoplásicas muestran un núcleo irregular, atípico, con citoplasma escaso y su inmunofenotipo incluye marcadores de células T, histiocitarios y de gránulos citotóxicos: CD2+, CD3e, CD3(+, CD5-, CD4-/+, CD8-/+, CD7-/+, CD16+, CD56+, CD57+; TIA-1+, perforina y granzima-B+. Con la hibridación in situ es posible demostrar la presencia de ARN nuclear del VEB (EBER) en la mayoría de los casos (48,49). No hay reordenamiento de los genes del TCR o cadenas pesadas de inmunoglobulinas (IgH) (3,5,13).
BIBLIOGRAFÍA
Willemze R, Beljaards RC, Meijer CJL. Classification of primary cutaneous T-cell lymphomas. Histopathology 1994; 24: 405-15.
Santucci M, Biggeri A, Feller A, Massi D, Burg G. Efficcy of histologic criteria for diagnosis of early mycosis fungoides. Am J Surg Pathol 2000; 24: 40-50.
Liu V, McKee PH. Cutaneous T-cell Lymphoprolipherative disorders: Approach for the surgical pathologist: Recent advances and clarification of confused issues. Advance Anat Pathol 2002; 2: 79-100.
Connors JM, Hsi ED, Foss FM. Lymphoma of the skin. Hematology (Am Soc Hematol Educ Program). 2002; 263-82.
Ralfkiaer E, Delsol G, Willemze R, Jaffe ES. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. En: Jaffe ES, Lee Harris N, Stein H, Vardiman JW, editores. WHO Classification. Tumours of Haematopoietic and Lymphoid Tissues. 1.ª ed. Lyon: IARC Press., 2001. p. 221-224.
Willemze R, Kerl H, Berti E y cols. EORTC classification for primary cutaneous lymphomas: A proposal from the cutaneous lymphoma study group of the European Organization for Research and Treatment of Cancer. Blood 1997; 1: 345-71.
Ploysangam T, Breneman DL, Mutasim DF. Cutaneous Pseudolymphomas. J Am Acad Dermatol 1998; 38: 877-95.
Rijlaarsdam JU, Scheffer E, Meijer JLM, Willemze R. Cutaneous Pseudo-T-Cell-Lymphomas. A clinicopathologic study of 20 patients. Cancer 1992; 69: 171-24.
Medeiros LJ, Picker LJ, Abel EA y cols. Cutaneous Lymphoid Hyperplaisa. Immunologic characteristics and assesment of criteria recently proposed as diagnsotic malignant lymphoma. J Am Acad Dermatol 1989; 21: 929-42.
Le Boit PE y Mc Calmont TH. En: Elder D, Elenitsas R, Jaworsky C, Jhonson B Jr, eds. Lever’s Histopathology of the skin, 8.ª ed. Philadelphia: Lippincott-Raven,1997. p. 805-846.
Kempf W, Dummer R, Burg G. Approach to lymphoproliferative infiltrates of the skin. The difficult lesions. Am J Clin Pathol 1999; 111(Suppl 1): S84-S93.
Wood GS, Hu CH. Parapsoriasis. En: Freedberg IM y cols. Fitzpatrick’s Dermatology in General Medicine. 5.ª ed. New York: McGraw Hill, 1999. p. 546-53.
Strutton G. Cutaneous infiltrates-lymphomatous and leukemic. En:Weedon D, editor. Skin Pathology, 2.ª ed. Londres: Churchill-Livingstone, 2002. p. 1095-1138.
King-Ismael D, Ackerman B. Guttate Parapsoriasis/Digitate Dermatosis (Small Plaque Parapsoriasis) is Mycosis Fungoides. Am J Dermatopathol 1992; 14: 518-30.
Kotz EA, Anderson D, Thiers BH. Cutaneous T-cell lymphoma. J Eur Acad Dermatol Venereol 2003; 17: 131-7.
Stevens RE, Ke MS, Birol A y cols. A simple clinical scoring system to improve the sensitivity and standarization of the diafnosis of mycosis fungoides type cutaneous T-cell lymphoma: logistic regression of clinical and laboratory data. Br J Dermatol 2003; 149: 513-22.
Smith NP. Hostologic criteria for early diagnosis of cutaneous T-cell lymphoma. Dermatol Clinics 1994; 12: 3153-22.
Glusac EJ. Criterion by criterion, Mycosis Fungoides. Am J Dermatopathol 2003; 25: 264-9.
Glusac EJ. Of cells and architecture: new approaches to old criteria in mycosis fungoides. J Cutan Pathol 2001; 28: 169-73.
Heald PW, Edelson RL. Lymphomas, Pseudolymphomas and related conditions. En: Freedberg IM y cols. Fitzpatrick’s Dermatology in General Medicine. 5.ª ed. New York: McGraw Hill, 1999. p. 1227-74.
Murphy GF, Elder DE. Non melanocytic tumors of the skin. AFIP. Bethesda 1991: 155-87.
Marti RM, Pujol RM, Servitje O y cols. Sézary Syndrome and related variants of classic cutaneous T-cell lymphoma. A descriptive and prognostic clinicopathologic study of 29 cases. Leukemia Lymphoma 2003; 44: 59-69.
Vonderheid EC, Bernengo MG, Burg G, Duvic M, Heald P, Laroche L, Olsen E, Pittelkow M, Russell-Jones R, Takigawa M, Willemze R. Update on erythrodermic cutaneous T-cell lymphoma: report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 2002; 46: 95-106.
Prince HM, O´Keefe R, McCormack C y cols. Cutaneous lymphomas; which pathological classification?. Pathology 2002; 34: 36-45.
Scarabello A, Leinweber B, Ardigó M y cols. Cutaneous lymphomas with prominent granulomatous reaction. A potential pitfall in the histopathologic diagnosis of cutaneous T- and B- cell lymphomas. Am J Surg Pathol 2002; 26: 1259-68.
Topar G, Zelger B, Schmuth M, Romani N, Thaler J, Sepp N. Granulomatous Slack Skin: A Distinct Disorder or a Variant of Mycosis Fungoides? Acta Derm Venereol 2001; 81: 42-44.
DeBloom J, Severson J, Gaspart A, Scott G. Follicular mycosis fungoides: a case report and review of the literature. J Cutan Pathol 2001; 28: 318-24.
Agnarsson BA, Kadin ME. Peripheral T-cell lymphomas in children and adolescents. Sem Diag Pathol 1995; 12: 314-24.
Fierro MT, Novelli M, Savoia P y cols. CD45RA+ immunophenotype in mycosis fungoides: clinical, histological and immunopehenotypical features in 22 patients. J Cutan Pathol 2001; 28: 356-62.
Holm N, Flaig MJ, Yazdi AS, Sander CA. The value of molecular analysis by PCR in the diagnosis of cutaneous lymphocytic infiltrates. J Cutan Pathol 2002 ;29: 447-52.
Dadej K, Gaboury L, Lamarre L y cols. The value of clonality in the diagnosis and follow-up of patients with cutaneous T-cell infiltrates. Diagn Mol Pathol 2001; 10: 78-88.
Jones D, Duvic M. The current state and future of clonality studies in mycosis fungoides. J Invest Dermatol 2003; 121(3): ix-x.
Le Boit PE. Lymphomatoid papulosis and cutaneous CD30+ lymphoma. Am J Dermatopathol 1996; 18: 221.
Stein H, Foss H-D, Dürkop H et al. CD30+ anaplastic large cell lypmhoma: a review of its histopathologic, genetic, and clinical features. Blood 2000; 96(12): 3681-3695.
Paulli M, Berti E, Rosso R, et al. CD30/Ki-1 positive lymphoproliferative disorders of the skin: clinicopathologic correlation and statistical analysis of 86 cases. A multicentric study from the European Organization for Research and Treatment of Cancer Lymphoma Project Group. J Clin Oncol 1995; 13: 1343-1354.
Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. CD30+ cutaneous lymphoproliferative disorders: the Stanford expirience in lymphomatoid papulosis and primary cutaneous anaplastic large cells lymphoma. J Am Acad Dermatol 2003; 49(6): 1049-58.
Kummer JA, Vermeer MH, Dukers D, et al. Most primary cutaneous CD30-positive lymphoproliferative disorders have a CD4-positive cytotoxic T-cell phenotype. J Invest Dermatol 1997; 109: 636-40.
Boulland ML, Wechsler J, Bagot M, Pulford K, Kanavaros P, Gaulard P. Primary CD30-positive cutaneous T-cell lymphomas and lymphomatoid papulosis frequently express cytotoxic proteins. Histopathology 2000; 36: 136-144.
Baylot-Barry M, Groppi A, Vergier B, Pulford K, Merlio JP. Characterization of t(2;5) reciprocal transcripts and genomic breakpoints in CD30+ cutaneous lymphoproliferations. Blood 1998; 91: 4668-76.
Sterry W, Jahn S. Other systemic lymphomas with skin infiltrations. En: Freedberg IM y cols. Fitzpatrick’s Dermatology in General Medicine. 5.ª ed. New York: McGraw Hill, 1999. p. 1250-58.
Ralfkiaer E, Stein H, Wantzin GL, Thomsen K, Ralfkiaer N, Mason DY. Lymphomatoid papulosis. Characterization of skin infiltrates by monoclonal antibodies. Am J Clin Pathol 1985; 84: 587-93.
Wood GS, Crooks CF, Uluer AZ. Lymphomatoid papulosis and associated cutaneous lymphoproliferative disorders exhibit a common clonal origin. J Invest Dermatol 1995; 105: 51-55.
Le Boit PE. Granulomatous slack skin. Dermatol Clin 1994; 12: 375-389.
Jaffe ES. Subcutaneous panniculitis-like T-cell lymphoma. En: Masen DY y Harris NL, editoores. Human lymphoma: clinical implications of the REAL classification. Londres: Springer; 1999. Cap. 34.
Wang CE, Su WP, Kurtin PJ. Subcutaneous panniculitic T-cell lymphoma. Int J Dermatol 35, 1: 1-8.
Munn SE, McGregor JM, Jones A, et al. Clinical and pathologic heterogeneity in cutaneous gamma-delta T-cell lymphoma: a report of three cases and a review of the literature. Br J Dermatol 1996; 135: 976-81.
Harris NL, Jaffe ES, Stein H y cols. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361-92.
Chan JKC, Jaffe ES, Ralfkiaer E. Extranodal NK/T-cell lymphoma, nasal type. En: Jaffe ES, Lee Harris N, Stein H, Vardiman JW, editores. WHO Classification. Tumours of Haematopoietic and Lymphoid Tissues. 1.ª ed. Lyon: IARC Press., 2001. p. 204-7.
Xu ZG, Iwatsuki K, Oyama N, Ohtsuka M, Satoh M, Kikuchi S, Akiba H, Kaneko F. The latency pattern of Epstein-Baar virus infection and viral IL-10 expression in cutaneous natural killer/T-cell lymphomas. Br J Cancer 2001; 84: 920-5.
![]()