
Vol. 42, n.º 2, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 2, 2009 |
CASUÍSTICA
Pere Huguet, Carme Dinarès, Carmela Iglesias, Carmen M.ª Blázquez, Felicia Marginean, Daniel Badia, Josep Castellví, Natàlia Tallada, Santiago Ramón y Cajal
«El que no sabe lo que busca
no entiende lo que encuentra»
Claude Bernard
Departament d’Anatomia Patológica. Hospital Universitari Vall d’Hebron. Universitat Autònoma de Barcelona. Barcelona.
RESUMEN
Para un patólogo preparado, reconocer un tumor cuando presenta la morfología característica y está en su localización habitual es fácil. Pero cuando se presenta en un lugar insólito y además la biopsia para diagnóstico es pequeña, es fácil caer en un error de orientación y, aunque a veces las técnicas auxiliares nos puedan ayudar, en algunas ocasiones no se concreta el diagnóstico hasta que una biopsia completa o la extirpación del tumor nos permiten ver la totalidad de la lesión, con la sobrecarga de tiempo, riesgo para el paciente y gasto económico que ello conlleva.
El Prof. Rosai describe esta situación con la historia que contaba Lauren V. Ackerman y que tituló «El hombre de Estambul»: el empecinamiento en no reconocer un tumor porque no está en «su» lugar.
Aportamos dos casos de sarcomas de partes blandas que se presentaron en localizaciones que podríamos llamar «invertidas»: un sarcoma sinovial mandibular y un mioepitelioma maligno yuxtaarticular en un dedo del pie y exponemos las dificultades que presentaron para su diagnóstico inicial.
Palabras clave: Sarcomas de partes blandas, sarcoma sinovial, mioepitelioma, diagnóstico, hombre de Estambul.
SUMMARY
A tumour with a characteristic morphological appearance occurring in a usual location is easy to diagnose. However, if a tumour is found in an unexpected site and only a small amount of biopsy material is available, it is more difficult to reach a correct diagnosis.
Dr Rosai quotes an expression often used by Lauren V. Ackerman to describe such a situation: «the man from Istanbul», ie. a common lesion occurring in the wrong place.
Two cases of common soft tissue tumours located in uncommon places are presented: a synovial sarcoma in the mandible and a malignant myoepithelioma (mixed tumour) near the phalangeal joint of the foot. The difficulties of a correct initial diagnosis are discussed.
Keywords: Soft tissue sarcomas, synovial sarcoma, myoepithelioma, diagnose, man from Istanbul.
INTRODUCCIÓN
–«Dr Livingstone, I presume»
Con esta frase, dicen, en 1871 el periodista galés Henry Stanley saludó al misionero y explorador escocés David Livingstone cuando lo encontró en Tanganika, en el corazón de África, después de buscarlo durante casi un año, sin extrañarse ni exteriorizar la más mínima emoción. Aunque no lo había visto nunca, lo había reconocido fácilmente a partir de la descripción que de él le habían hecho en su periódico cuando le encargaron la misión y porque lo estaba buscando en el lugar adecuado (1).
El Prof Juan Rosai cuenta siempre la historia del «hombre de Estambul» (2) que, dice, explicaba el Prof Lauren V. Ackerman para ilustrar la dificultad de reconocer un elemento cuando no se halla en su entorno habitual, y que yo le he oído explicar a él en más de un par de ocasiones. Podríamos resumirla así:
«Había un hombre llamado John que vivía en un apartamento en Nueva York. Cada mañana, al abrir la puerta y salir a trabajar, se encontraba con su vecino Fred que salía, a la misma hora, de la puerta de enfrente.
–Buenos días, Fred. Decía.
–Buenos días, John. Contestaba Fred. Y cada uno se iba hacia su trabajo.
Un día, John tuvo que viajar, por cuestión de negocios, a Estambul. Entre el viaje movido y los nervios por ser su primera salida al extranjero, llegó cansado. Se fue directo al hotel y se echó a dormir. Por la mañana se levantó, se arregló y salió de la habitación. Al abrir la puerta, de la habitación de enfrente salió Fred en el mismo momento. Tal vez alguna cosa en el subconsciente de John le dijo que Fred estaba ahí, como cada mañana en los últimos muchos años pasados como vecinos. Pero el cerebro estructurado de John le hizo ver que se encontraba en Estambul y que dicha posibilidad, por tanto, era prácticamente impensable. Y John no la admitió. Sencillamente, dio media vuelta y siguió por el pasillo hacia su cita de trabajo.
Pero, realmente, Fred, su vecino, se hallaba ese mismo día en Estambul por una cuestión de negocios.»
Presentamos dos tumores que por sus características morfológicas no nos habrían presentado problemas en el diagnóstico (ayudándonos de técnicas auxiliares, seguramente) a no ser porque se presentaron en lugares «equivocados» y, tal vez, cambiados: uno en el ángulo mandibular y el otro en un dedo del pie. Debe añadirse, además, la dificultad asociada de una biopsia tru-cut, pequeña, en ambos casos, siempre difícil de valorar en los tumores de partes blandas.
PRESENTACIÓN DE LOS CASOS
Caso 1
Varón de 33 años que acudió a la consulta de Cirugía Máxilofacial con una lesión tumoral en trígono retromolar derecho, ulcerada y mal definida, que infiltraba hueso mandibular. Con la sospecha clínica de carcinoma escamoso se le practicó una PAAF para establecer el diagnóstico: se observaba un fondo necrótico-hemorrágico con células atípicas, de citoplasma poco aparente y núcleos redondeados o fusiformes, aisladas o formando grupos y placas variables, a veces con tendencia a formar haces. Se acompañaban de abundante matriz mixoide fibrilar (fig. 1). En el bloque celular predominaba la necrosis con algunas células atípicas fusiformes y componente vascular acompañante.
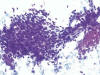
Fig. 1.
Al mismo tiempo se tomó una pequeña muestra para biopsia que mostraba abundante material necrótico y grupos densos de células con moderada atipia, de patrón sólido, con citoplasmas claros o vesiculosos y núcleos redondeados, con mitosis aisladas, estroma mixoide y alguna microcalcificación; no existían áreas evidentes de distribución fasciculada (fig. 2). Dada la localización y tratándose de un paciente joven se pensó en la posibilidad de tumores odontogénicos con células claras y fusiformes y estroma mixoide, tumores de glándula salival con las mismas características y, según parecía indicar la citologia, sarcomas fusocelulares con estroma mixoide. Se practicó IHQ que mostró positividad para vimentina, citoqueratina 7, EMA y Bcl-2. Fueron negativos los marcadores neurales y melánicos, los musculares, CD99, CD34, y pan-citoqueratina. Con el diagnóstico de sospecha de sarcoma sinovial se procedió, mediante FISH, a determinar la traslocación del gen SYT (18q11) que fue positiva.
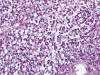
Fig. 2.
Poco después se nos remitió la hemimandibulectomía derecha que en el ángulo mandibular presentaba una tumoración de 8 x 8 cm que rodeaba toda la mandíbula infiltrando partes blandas y mucosa de encía y trígono que estaba ulcerada. Al corte mostraba áreas de necrosis, calcificación central y zonas tumorales de aspecto semidenso con focos mixoides, de color blanco-grisáceo (figs. 3 y 4) infiltrando y destruyendo el hueso. La HE mostró un patrón característico de sarcoma sinovial monofásico: las células, en su mayoría, tenían hábito fusiforme y formaban fascículos densos con atipias y mitosis atípicas evidentes (fig. 5); en algunas zonas mostraban el aspecto redondeado y claro que se había visto en la biopsia y en algunos escasos focos presentaban aspecto aún más indiferenciado con presencia de células hialinas (fig. 6). No se observó componente bifásico. El estroma mixoide era abundante en toda la tumoración, así como la necrosis. No se observaron restos de epitelio odontogénico en ninguno de los cortes examinados ni se detectó displasia en la mucosa residual conservada para descartar un posible carcinoma sarcomatoide inicial.
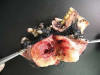
Fig. 3.

Fig. 4.
Fig. 5.

Fig. 6.
Caso 2
Varón de 25 años que acudió a nuestro Hospital por la ulceración de una tumoración en la parte distal y ventral del segundo dedo del pie izquierdo, de 4 meses de evolución, con dolor creciente. Se trataba de una masa de unos 4 ó 5 cm de diámetro, ulcerada, de consistencia densa. La RM mostró una tumoración de partes blandas posiblemente relacionada con el tendón del flexor y se sugirió el diagnóstico de tumor de células gigantes de la vaina del mismo, si bien no estaba clara la relación con la 3.ª falange. Se practicó biopsia tru-cut que mostró un tumor constituido en su mayor parte por células de aspecto plasmacitoide, de citoplasma acidófilo denso y núcleo redondeado u oval, excéntrico, con moderada atípia y con algunas mitosis atípicas aisladas. Se disponían en cordones irregulares o en grupos de aspecto alveolar con escasa cohesión celular y con centros vacíos o mixoides (fig. 7). En alguna zona los grupos eran más sólidos y el aspecto epitelioide de las células algo más acusado. El estroma entre los grupos era colágeno con zonas de aspecto hialinizado y en zonas más laxo y también presentaba degeneración mixoide evidente (fig. 8). No se observaba componente inflamatorio acompañante ni áreas de necrosis. La tinción de PAS y digestión con diastasa mostró presencia de glucógeno en la mayoría de las células.
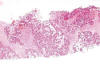
Fig. 7.
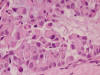
Fig. 8.
Con este patrón morfológico y dadas la edad del paciente y la localización de la lesión se pensó en un posible rabdomiosarcoma alveolar, un tumor rabdoide extrarrenal o un sarcoma sinovial pobremente diferenciado. Aunque no correspondía por la edad, se consideraron también el condrosarcoma mixoide y el tumor fibromixoide osificante. Se solicitaron marcadores IHQ que mostraron expresión intensa y difusa de vimentina, pan-citoqueratina, EMA, CK7 y proteína S100 en la mayoría de células. Fueron negativas las actinas de músculo liso y total, desmina, miogenina y mio-D, así como Bcl-2 y los marcadores neurales y melánicos. El estudio de la traslocación del gen SYT (18q11) por FISH fue negativo.
Se estableció un diagnóstico provisional de «tumor maligno de partes blandas de origen no filiado» sugiriéndose la extirpación completa para su valoración definitiva. Pocos días después se practicó la desarticulación metatarsofalángica del dedo (fig. 9).
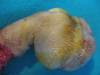
Fig. 9.
Se nos remitió la pieza que presentaba la masa descrita. Al corte el tumor medía 3,5 cm de diámetro mayor, estaba bien delimitado y ocupaba dermis e hipodermis del pulpejo del dedo infiltrando epidermis y no afectaba, aparentemente, ni hueso ni tendón. Tenía consistencia densa algo elástica, color pardo claro y aspecto sólido con zonas mixoides (fig. 10).
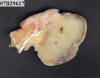
Fig. 10.
El estudio con HE de la sección del tumor, a baja resolución, mostró la imagen típica de un tumor mixto de glándula salival de predominio mioepitelial. El estroma mixoide era abundante y en algunas zonas francamente condromixoide (fig. 11). Las células mioepiteliales eran en su mayoría de tipo hialino o plasmacitoide, en algun punto eran más sólidas y epitelioides con leve diferenciación ductal, y mostraban atipias y mitosis atípicas aisladas (fig. 12). No se observó relación con el hueso ni con el tendón. Aunque no estaba encapsulado, los límites del tumor eran netos y no se observó invasión vascular ni perineural. No se constató necrosis intratumoral. El perfil IHQ se completó con calponina y P63, positivas. La proteína glial fue negativa. Se estableció el diagnóstico definitivo de Mioepitelioma de partes blandas con diferenciación glandular focal (tipo tumor mixto), con transformación maligna «in situ».
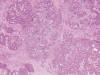
Fig. 11.
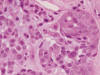
Fig. 12.
COMENTARIOS
El sarcoma sinovial (SS) es un tumor de partes blandas que, a pesar de su nombre, no tiene relación con el tejido sinovial, si bien se sitúa cerca de articulaciones y en algunos casos se ha descrito su localización intraarticular (3,4). Representa el cuarto sarcoma en frecuencia en partes blandas (entre el 5 y el 14,5%). Predomina en extremidades y es poco frecuente en cabeza y cuello (9%) donde se han publicado unos 75 casos, generalmente en región retro y parafaríngea y área del esternocleidomastoideo, y solo 3 primarios mandibulares (5,6). En nuestro caso el tumor afectaba hueso y partes blandas y no podemos saber si su inicio fue central o periférico.
El SS suele crecer lentamente en forma de masa indolora y, en ocasiones, presenta calcificaciones que se detectan radiológicamente. Histológicamente se describen tres tipos: patrón bifásico, con componente epitelial y mesenquimal en proporción variable, monofásico epitelial o monofásico mesenquimal, éste con células fusiformes formando haces alargados densos o dispuestas en sábana, con núcleos que se superponen, a veces formando empalizadas, escasa atipia y algunas mitosis. La necrosis tumoral es variable. El estroma suele ser colágeno y presentar vasos de aspecto hemangiopericitoide. Pocas veces se describe estroma mixoide y en escasas ocasiones las células son redondas o claras. En los SS poco diferenciados que describió Enzinger suele encontrarse un patrón con células hialinas y el comportamiento es más agresivo (7).
La IHQ, aunque no específica, ayuda a su diagnóstico y al diagnóstico diferencial. Las células epiteliales del SS bifásico expresan citoqueratinas, especialmente 7 y 19, y EMA. Las fusiformes pueden también expresar estos marcadores, además de vimentina, Bcl-2 y, en ocasiones CD99. CD34 es, en general negativo, si bien se ha descrito expresión en algún caso de SS poco diferenciado (6).
La definición de la t(X;18) mediante FISH confirma el diagnóstico en el 90% de los SS (7).
Los tumores que se parecen a los de la glándula salival se han descrito en numerosas partes del cuerpo, relacionándolos, probablemente, con estructuras glandulares exocrinas de estas zonas. Con algunas dificultades añadidas se han estudiado también en mama y pulmón, donde no se piensa tan a menudo en esta relación (8) .Y si los situamos en partes blandas profundas la poca frecuencia los convierte en tumores casi exóticos. Los mioepiteliomas de partes blandas son tumores que hasta la última edición de la OMS no han sido reconocidos o bien caracterizados (9). Anteriormente se les consideraba una forma de presentación profunda de tumores anexiales cutáneos o se les confundía con los paracordomas descritos en 1951. Sus características morfológicas y su perfil inmunohistoquímico característico permite identificarlos con facilidad. Las células mioepiteliales presentan un espectro de diferenciación amplio, pudiendo mostrar hábito epitelioide, halino o plasmacitoide, fusiforme o de células claras. En los tumores de partes blandas suele predominar el patrón epitelioide con alguna diferenciación ductal (si éste es predominante, hablaremos ya de tumor mixto). La IHQ pone de manifiesto la expresión casi constante de citoqueratinas, EMA, vimentina, proteína S-100, proteína glial, calponina y P-63. La expresión de actina de músculo liso que se observa en las células mioepiteliales normales suele perderse en los tumores, más aún en los malignos (10) Las mitosis suelen ser escasas y el pleomorfismo nuclear mínimo, pero se han descrito hasta un 30 % de casos malignos, en cuyo caso suele haber también necrosis intratumoral (11). El estroma es condromixoide con mayor o menor hialinización. Su semejanza morfológica y, en parte, inmunohistoquímica, con el tumor fibromixoide osificante, ha llevado en algunos casos a estudiar el posible parentesco entre ambos, aunque sin resultados definitivos (12).
Son tumores que predominan en gente joven, incluso en la infancia (13) y que suelen localizarse en el tejido subcutáneo o subfascial de las extremidades. En algún caso se han descrito en hueso. No se han recogido más de 150 casos en la literatura, y solo en algunos se han estudiado sus características genéticas, sin llegar a ninguna conclusión por ahora. Aunque la mayoría son benignos, los casos malignos tienden a la recurrencia y pueden dar metástasis y provocar la muerte del paciente (8).
Todos sabemos que el reconocimiento de algo es más fácil si este algo se presenta en su contexto habitual. Como explicaba Rosai (2) en la historia que nos ha servido de introducción, los patólogos nos hemos encontrado frecuentemente en situaciones como las de los dos casos aquí expuestos. Una lesión que por si misma podría ser de fácil e inmediato reconocimiento si estuviera en su contexto, ve dificultado su diagnóstico porque no pensamos que «pueda aparecer ahí» (donde no toca). Y si además la biopsia es de pequeño tamaño la dificultad aumenta al tratarse de tumores de partes blandas, donde el patrón de una pequeña parte del tumor puede no ser en absoluto representativa del resto de la masa.
En los dos casos presentados la pequeña biopsia para diagnóstico no era demostrativa de la mayor parte del tumor, si bien en el primero la presencia de algunos haces de células fusiformes en la PAAF y un perfil IHQ característico orientaron hacia el diagnóstico correcto que se confirmó con el FISH. En la segunda biopsia fue necesario ver la totalidad del tumor para reconocer su identidad definitiva, confirmándose después mediante la IHQ. En ninguno de los dos casos hemos tenido noticias de recidivas o metástasis 1 año y 2 años después de sus respectivas exéresis.
Es útil equivocarse y es práctico aprender de los errores (propios), especialmente cuando pueden enmendarse en poco tiempo y con mínimo riesgo para el paciente.
Como decía el Prof Juan Rosai al concluir su relato «creo que hay dos clases de patólogos para los que la historia de Ackerman no tiene sentido: los que trabajan en Estambul y los que no habrían reconocido a Fred ni en Nueva York» (2).
BIBLIOGRAFÍA
Moret X. Exploradors del Nil. El Periódico de Catalunya. Dominical. Marzo 2008.
Rosai J. Dr Lauren V. Ackerman and his man from Istanbul. Semin Diagn Pathol 2003; 20: 247-8.
Namba Y, Kawai A, Naito N, Morimoto Y, Hanakawa S, Inoue H. Intraarticular synovial sarcoma confirmed by SYT-SSX fusion transcript. Clin Orthop 2002; 395: 221-6.
Sheldon PJ, Forrester DM, Learch TJ. Imaging of intraarticular masses. RadioGraphics 2005; 25: 105-19.
Meizoso T, De Alava E, Garrido-Ruiz M, Enguita AB, Martínez-González MA, Ballestín C. Sarcoma sinovial intramandibular. A propósito de un caso. Rev Esp Patol 2007; 40: 188-91.
Koga C, Harada H, Kusukawa J, Kameyama T. Synovial sarcoma arising in the mandibular bone. Oral Oncology 2005; 41: 45-8.
Fisher C, De Bruijn DRH, Geurts van Kessel A. Synovial Sarcoma. In Fletcher CDM, Unni KK, Mertens F WHO Classification of tumours. Pathology and Genetics. Tumours of Soft Tissue and Bone. Lyon 2002. Pp 200-4.
Bennet AK, Mills SE, Wick MR. Salivary-type neoplasms of the breast and lung. Semin Diagn Pathol 2003; 20: 279-304.
Kilpatrick SE, Limon J. Mixed Tumour/Myoepithelioma/ Parachordoma. In Fletcher CDM, Unni KK, Mertens F WHO Classification of tumours. Pathology and Genetics. Tumours of Soft Tissue and Bone. Lyon 2002. Pp. 198-9.
Alos L, Cardesa A, Bombi JA, Mallofre C, Cuchi A, Traserra J. «Myoepithelial tumors of salivary glands; a clinicopathologic, immunohistochemical, ultrastructural and flow-cytometric study. Semin Diagn Pathol 1996; 13: 138-47.
Lee JR, Georgi DE, Wang BY. Malignant myoepithelial tumor of soft tissue: a report of two cases of the lower extremity and a review of the literature. Ann Diagn Pathol 2007; 11: 190-8.
Min KW, Seo IS, Pitha J. Ossifying fibromyxoid tumor: modified myoepithelial cell tumor? Report of three cases with immunohistochemical and electron microscopic studies. Ultrastruct Pathol 2005; 29: 535-48.
Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol 2007; 31: 1813-24.
![]()