
Vol. 42, n.º 3, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 3, 2009 |
CASUÍSTICA
Bernardo Weil Lara
Servicio de Anatomía Patológica. Hospital
Materno-Infantil. Avenida Arroyo los Ángeles, s/n. Málaga 2011.
Bernardo.weil.sspa@juntadeandalucia.es
RESUMEN
El glioma angiocéntrico se incluye como una nueva entidad en la última clasificación de la OMS dentro la categoría «otros tumores neuroepiteliales» y considerado grado 1. Presentamos un caso en un niño de 10 años con una neoplasia cerebral hemisférica que se manifestó clínicamente con crisis epilépticas. Radiológicamente la tumoración era hiperintensa en T2. Histológicamente presentaba, como rasgos distintivos, células monomorfas bipolares fusiformes con patrón angiocéntrico, y pseudorrosetas radiales. Las técnicas de inmunohistoquímica mostraban expresión para PGFA, y una característica positividad para EMA en forma de gotas citoplasmáticas o formaciones tubulares microscópicas. La neoplasia fue extirpada en su totalidad no detectándose recidivas en controles radiológicos, en seis años de seguimiento.
Palabras clave: Glioma angiocéntrico, diferenciación ependimal.
SUMMARY
The latest WHO classification included angiocentric glioma as a new entity in the category of «other neuroepithelial tumours» and considered it a grade 1 tumour. We present a case in a 10 year old boy with a brain tumour clinically manifested as seizures. Radiologically the tumour was T2-hyperintense. Histologically, distinctive features were monomorphous bipolar spindle cells in an angiocentric pattern, and radial pseudorosettes. The immunohistochemical study showed expression for GFAP, and a characteristic positivity for EMA as dot-like or cytoplasmic tubular microscopic formations. The tumour was excised as a whole and no recurrences were detected in six years of follow-up with imaging techniques.
Keywords: Angiocentric glioma, ependymal differentiation.
INTRODUCCIÓN
El glioma angiocéntrico se considera una neoplasia cerebral estable o de crecimiento lento, asociada a epilepsia, que afecta a niños y a adultos jóvenes. Esta neoplasia se caracteriza histológicamente por presentar un patrón de crecimiento angiocéntrico, y estar constituida por células monomorfas bipolares con características inmunohistoquímicas y ultraestructurales de diferenciación ependimaria. Se trata de una lesión recientemente descrita con una relación incierta con otras neoplasias de diferenciación ependimaria. La incidencia no se conoce todavía. Se presenta un caso y se revisa la literatura sobre el tema.
DESCRIPCIÓN DEL CASO
Niño de 10 años que ingresó por presentar crisis epilépticas en tres ocasiones en el mismo día. Una semana antes presentó episodios breves de «reacción de parada» (cese de actividad con fijación de la mirada) sin desconexión de su entorno. Las crisis no respondieron a tratamiento. Se detectó una tumoración cerebral localizada en lóbulo frontal izquierdo que anatómicamente incluía parte anterior del lóbulo parietal, la cual llegaba hasta la cisura interhemisférica anterior y, superiormente, con la sustancia gris cortical de la convexidad cerebral. Dicha lesión presentaba en T1 baja señal y en T2 densidad protónica alta. En secuencia T2 coronal la hiperdensidad en sustancia blanca parecía extenderse hacia nivel periventricular (asta frontal izquierda). Tras la inyección de contraste parecía presentar una muy discreta captación en la periferia de la lesión en T1. Se trataba de una lesión en parte sólida y, el resto, peor definido en sustancia blanca con aspecto digitiforme. Las dimensiones máximas eran de 4 x 3 cms aproximadamente. Se realizó extirpación quirúrgica.
En la evolución del paciente, desaparecen las crisis tras la intervención. Al año y medio reinicia las crisis en número de 1-4/día en forma de desconexión del medio, de segundos de duración, seguidas de automatismos verbales y sin sueño post-ictal. Las crisis, que respondieron temporalmente a diferentes tratamientos administrados en un periodo de cinco años, se hacen progresivamente resistentes con los fármacos antiepilépticos disponibles, lo que era compatible clínicamente con epilepsia focal sintomática, probablemente relacionada con área de gliosis postquirúrgica.
En diferentes estudios de RMN realizados durante seis años de evolución no se ha detectado ninguna recidiva tumoral.
HALLAZGOS ANATOMO-PATOLÓGICOS
La pieza correspondía a cuatro fragmentos de entre 1,2 y 3,3 cm. La tumoración ocupaba la corteza cerebral y sustancia blanca subcortical (fig. 1). En la zona más sólida se evidencian abundantes elementos fibrilares, en los que podían apreciarse en nuestro caso focos de microhemorragias así como algunos vasos esclerosados y presencia focal de macrófagos hemosiderínicos. Alrededor de estos vasos de paredes esclerosadas se disponían abundantes acúmulos celulares constituyendo fascículos o nódulos. Se pudo observar focalmente un patrón microquístico (fig. 2). Una destacada característica, particularmente llamativa en las zonas de infiltración subcorticales, era la formación de estructuras constituidas por células monomorfas bipolares fusocelulales, orientadas en relación a vasos corticales de todos los calibres, en monocapa o multicapa, a lo largo de los ejes vasculares así como abundantes pseudorrosetas radiales (fig. 3). La orientación circunferencial o longitudinal de los vasos predominaba en las áreas menos celulares (fig. 4) mientras que las pseudorrosetas radiales prevalecían en las regiones más celulares (fig. 5). Se identificaron neuronas incluidas en la tumoración. No se observaron mitosis, proliferaciones microvasculares complejas ni necrosis. Tampoco calcificaciones, fibras de Rosenthal, cuerpos granulares eosinófilos. No se observó, en este caso, infiltración subpial. La neoplasia fue diagnosticada inicialmente, en base a las características citológicas fusocelulares, de astrocitoma grado 2, al igual que otros casos incluidos en las series que describen la entidad.
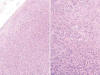
Fig. 1:
Panorámica de la corteza cerebral en la que destaca la zona más celular de la
neoplasia (HE x100).
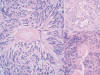
Fig. 2:
Patrón fascicular-nodular en torno a vasos esclerosados. Detalle de un foco
microquístico (esquina superior derecha) (HE x400).
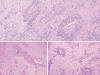
Fig. 3:
Predomina el patrón angiocéntrico en las áreas más periféricas (HE).
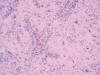
Fig. 4:
Observe el característico patrón angiocéntrico lineal. Se pueden encontrar
neuronas, probablemente atrapadas (HE x400).
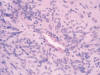
Fig. 5:
Detalle de las pseudorrosetas perivasculares constituidas por células fusiformes
bipolares (HE x400).
Con el objetivo de catalogar la neoplasia a la luz de la nueva clasificación de la OMS, se realizaron técnicas de inmunohistoquímica con los siguientes anticuerpos comerciales prediluidos (DAKO): PGFA, EMA, Vimentina, Sinaptofisina, S-100, Cromogranina, Enolasa, Neurofilamentos, CD99 y CD34. Las células neoplásicas mostraron expresión intensa para PGFA, CD99 (fig. 6) y Vimentina (fig. 7). Destaca la positividad para el EMA, en forma de gotas citoplasmáticas o formaciones tubulares microscópicas (fig. 8). El Ki67 fue <1%. Fueron negativos en las células neoplásicas en este caso la S-100, Enolasa, Neurofilamentos, Sinaptofisina y Cromogranina. El CD34 sólo se expresaba a nivel vascular.
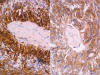
Fig. 6:
Positividad para la PGFA y para el CD99 en las células neoplásicas (HE x400).

Fig. 7:
Con Vimentina se puede evidenciar el patrón histológico de infiltración
angiocéntrico en la sustancia blanca subcortical (HE x200).
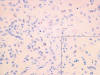
Fig. 8:
EMA. Observe el característico punteado citoplásmástico y en detalle las
formaciones anulares (HE x400).
DISCUSIÓN
Existen dos artículos en 2005 (1,2) que describen la entidad y uno en 2008 que aporta datos genéticos (3). En conjunto se describen 26 pacientes de edades comprendidas entre 2,3 y 70 años. Es típica la localización cerebrocortical superficial. Clínicamente esta neoplasia se caracteriza por presentarse como epilepsia siendo característica la epilepsia parcial crónica intratable. Con RMN y secuencia FLAIR son lesiones corticales bien delimitadas sólidas hiperintensas que no se realzan con contraste y que, usualmente, se extienden en la sustancia blanca subcortical (3). Las imágenes radiológicas que muestran extensión en forma de tallo al ventrículo adyacente, hiperintensas en T2 y FLAIR, se consideran diagnósticas (1). Las imágenes secuenciales indican que son neoplasias estables y de crecimiento muy lento (2).
Una característica común es la formación de estructuras constituidas por células monomorfas bipolares fusocelulares, orientadas en relación a vasos corticales de todos los calibres, en monocapa o multicapa, que se extienden a lo largo de los ejes vasculares y por pseudorrosetas radiales de apariencia ependimaria. Estas células, con frecuencia, se agregan bajo la pía-aracnoides de forma horizontal o perpendicular. Los núcleos son elongados y con punteado cromatínico granular. Algunos casos muestran regiones de crecimiento sólido, con más abundantes elementos fibrilares en forma de nódulos compactos tipo neurilemoma, así como células epitelioides en nidos y sábanas interrumpidas por hendiduras irregulares o cavidades. Estas células pueden contener estructuras paranucleares redondeadas u ovaladas, eosinofílicas, correspondientes a las microluces evidenciadas con el EMA. Las neuronas incluidas en la neoplasia se interpretan como atrapadas (2) o posiblemente intrínsecas a la lesión (1). Las mitosis son inaparentes o raras y no se observan proliferaciones microvasculares complejas ni necrosis. Se ha descrito, sin embargo, un caso recurrente, mitóticamente activo, anaplásico (2).
Las células son reactivas para PGFA, S-100, y Vimentina y no expresan antígenos neuronales. Las características ependimarias se evidencian con EMA en forma de gotas y microluces citoplasmáticas. Con microscopía electrónica se observan microluces con microvilli y cilios. El Ki-67 es del 1% o menos en la mayoría de los casos. En el caso anaplásico recurrente pasó del 1% a 10% (2). Desde el punto de vista genético, utilizando hibridación genómica comparativa (CGH), un caso presentó una pérdida en 6q24-q25. Asimismo, con CGH de alta resolución, se ha descrito un caso con una ganancia en el número de copias en 11p11.2, conteniendo el gen del receptor de la proteína tirosina fosfatasa tipo J (3). Se ha sugerido que esas neoplasias derivan de la glía radial bipolar (1). Son neoplasias estables siendo la excisión generalmente curativa. Se describió un caso en el que la radioterapia, sin resección quirúrgica, produce un posible efecto antiepileptogénico (3). El caso anaplásico recurrente afectó a un adulto en el que la tumoración fue resecada de forma subtotal (3). Al ser una entidad reciente existen pocos casos descritos en la literatura. Un resumen de un congreso que aporta dos casos de glioma angiocéntrico anaplásico en adultos, siendo uno de ellos una recurrencia a los 12 años (4). Otro resumen describe 3 casos pediátricos siendo uno de 4 años a nivel frontal con la peculiaridad de presentar 6 mitosis por 10 CGA pero libre de recurrencia a los 6 años (5).
En la última clasificación de la OMS se comenta la relación incierta de la entidad con otras neoplasias con diferenciación ependimaria (6). Autores como Lum y cols (7) y Lehman –que previamente había descrito un caso de ependimoma de localización cortical– (8,9) plantean, por las similitudes histológicas y el comportamiento biológico, que el glioma angiocéntrico puede estar relacionado con el ependimoma cortical. Es de destacar que el patrón de infiltración angiocéntrico es un dato fundamental en el diagnóstico del glioma angiocéntrico. Este patrón se describe en algunos artículos de ependimoma cortical (9) pero en otros no parece ser un dato relevante o bien no se ha tenido en cuenta en la descripción. La lesión ependimaria cortical se define en algunos casos como ependimoma grado II (10). En un artículo, en el que se revisan 14 casos de ependimomas supratentoriales corticales, se señala que dos casos fueron diagnosticados de ependimomas de células claras, uno de ependimoma celular y cuatro como ependimomas anaplásicos. De los 14 casos destaca que 6 fueron pediátricos (11). Es posible por tanto que a nivel cortical y subcortical se pueda observar un espectro de lesiones con diferenciación ependimaria. El Glioma angiocéntrico posee características clínicas, radiológicas y patológicas características que permite realizar un diagnóstico diferencial adecuado con otras entidades. Se consideran actualmente neoplasias de grado 1. No obstante es de destacar que se describen tres casos anaplásicos en adultos. Encontramos un caso descrito por Lehman similar al nuestro (caso nº3) en el que la extirpación eliminó los ataques epilépticos pero volvieron aproximadamente al año. El paciente en este caso rehusó una nueva intervención y fue perdido en el seguimiento (9).
BIBLIOGRAFÍA
Lellouch-Tubiana A, Boddaert N, Bourgeois M, et al. Angiocentric neuroepithelial tumor (ANET): a new epilepsy-related clinicopathological entity with distinctive MRI. Brain Pathol 2005; 15: 281-6.
Wang M, Tihan T, Rojiani AM, et al. Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J Neuropathol Exp Neurol 2005; 64: 875-81.
Preusser M, Hoischen A, Novak K, et al. Angiocentric glioma. Report of clinico-pathologic and genetic findings in 8 cases. Am J Surg Pathol 2008; 31: 1709-18.
Donev K, Scheithaur B, Ketterling R. Anaplastic angiocentric glioma: a report of two cases. FASEB J 2008; 22: 706.4.
Li J, Langford L, Adesina A, et al. Angiocentric glioma: elevated proliferation rate at clinical presentation does not preclude extended recurrence-free survival. FASEB J 2008; 22: 706.6.
Burger Pc, Rosenblum MK, Jouvet A, et al. Angiocentric glioma. En: Louis DN, Oggki H, Wiestler OD, et al, eds. World Health Organization Classification of tumours. Pathology and genetics of Tumours of the Nervous System. Lyon: IARC; 2007: 92-93.
Lum DJ, Halliday W, Watson M, et al. Cortical epndymoma or monomorphous angiocentric glioma? Neuropathology 2008; 28: 81-6.
Lehman NL, Jorden MA, Huhn S, et al. Cortical ependymoma. Pediatric Neurosurgery. 2003; 39: 50-4.
Lehman NL. Patterns of brain infiltration and secondary structure formation in supratentorial ependymal tumors. J Neuropathol Exp Neurol 2008; 9: 900-10.
Roncaroli F, Consales A, Fioravanti A, et al. Supratentorial cortical ependymoma: report of three cases. Neurosurgery 2005; 57: E192.
Miyazawa T, Hirose T, Nakanishi K, et al. Supratentorial ectopic cortical ependymoma occuring with intratumoral hemorrhage. Brain Tumor Pathol 2007; 24: 35-40.
![]()