
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
Eduardo Salido Ruiz
Dept. Anatomía Patológica y Laboratorio Unidad Investigación. Facultad de Medicina y Hospital Universitario de Canarias. Campus de Ofra. Tenerife
Los avances de la última década en el conocimiento de las bases moleculares de las enfermedades tienen importantes implicaciones en la patología renal, similar a otras áreas de la Medicina. El diagnóstico etiológico y el entendimiento de los mecanismos de la enfermedad son las categorías en que dichos avances han sido más definitivos. Es lógico pensar que estos avances se traducirán también en el futuro en avances terapéuticos, pero es el diagnóstico molecular lo que en la actualidad constituye una herramienta consolidada, y el objetivo de esta revisión.
Existen cuatro áreas principales de aplicación de la perspectiva molecular en nefropatología:
1. Diagnóstico etiológico de enfermedades hereditarias
2. Diagnóstico etiológico de enfermedades infecciosas
3. Perfil molecular de neoplasias
4. Perfil molecular de inflamaciones renales, incluyendo la patología del trasplante
El objetivo de esta revisión no es abarcar todas las enfermedades descritas, sino presentar una visión panorámica de estas aplicaciones moleculares, dando más detalles de algunos ejemplos representativos de cada grupo.
La interacción entre genes y medio ambiente matiza cualquier clasificación que consideremos para abordar esta revisión. En un extremo del espectro, podemos hablar de enfermedades de herencia mendeliana, debidas a mutaciones en un solo gen, y de determinismo sencillo: heredar un alelo mutado de una enfermedad dominante, o los dos alelos mutados para una recesiva, determina en gran medida el desarrollo de la enfermedad. En el otro extremo, las enfermedades causadas por agentes ambientales, como los virus, suelen afectar a un amplio rango de individuos, de distinto fondo genético, pero cada vez conocemos mejor la existencia de genes de susceptibilidad/resistencia a infecciones, que determinan la susceptibilidad individual a los mismos. Notablemente, la porción más ancha del espectro, la zona intermedia, esta ocupada por enfermedades multifactoriales, de determinismo complejo, en que heredar varios alelos de genes diferentes proporciona un riesgo genético, o predisposición individual al desarrollo de una enfermedad que sólo se manifiesta si concurren ciertas condiciones ambientales.
En toda esta gama de enfermedades, se puede definir en la actualidad los cambios en moléculas de alto contenido informativo asociados a las mismas. Los métodos diagnósticos moleculares apropiados a cada caso pueden ser:
Directos: cuando se busca el cambio molecular concreto asociado a la enfermedad (análisis mutacional) (1).
Indirectos: cuando se estudia cómo se han heredado marcadores moleculares ligados a la enfermedad (análisis de ligamiento) (2).
Monogénicos: estudio de cambios importantes (mutaciones patológicas) en un solo gen asociado a la enfermedad.
Multifactoriales: análisis de cambios sutiles (polimorfismos funcionales) en genes de susceptibilidad.
Detección de DNA, RNA o proteína, en función de la molécula más informativa y la disponibilidad de muestra (fig. 1) Evidentemente, todas las alteraciones genéticas pueden ser estudiadas en DNA, y todos los cambios hereditarios pueden ser evaluados a partir de cualquier célula nucleada. No obstante, la complejidad de muchos genes humanos y la heterogeneidad genética (mutaciones en varios genes diferentes pueden causar una misma enfermedad) y alélica (muchas mutaciones diferentes en un gen pueden causar la misma enfermedad) hace que con frecuencia sea más eficaz el estudio de mRNA o proteína. Una limitación fundamental del estudio de mRNA y proteínas es la necesidad de partir de muestras de tejido donde se exprese el gen de interés.

Fig. 1. Diferentes estrategias de abordaje molecular del diagnóstico.
Con frecuencia nos basamos en un análisis del DNA, mientras que en otros
casos puede ser más rentable un estudio basado en mRNA o proteína
codificada por el gen en cuestión.
ENFERMEDADES HEREDITARIAS
Con el descubrimiento de numerosos genes y variantes alélicas de los mismos que juegan un papel importante en la etiología y patogénesis de las enfermedades renales, se ha renovado el interés en el diagnóstico genético molecular. Con la excepción de la enfermedad poliquística renal autosómica dominante (ADPKD), las enfermedades hereditarias monogénicas que conllevan insuficiencia renal terminal son raras individualmente. No obstante, como grupo, la morbimortalidad de estas enfermedades y su impacto socioeconómico es alto. La tabla I recoge las entidades principales, de las cuales comentaremos en esta revisión sólo un par de ejemplos relativamente frecuentes, y una enfermedad rara, hiperoxaluria primaria, de particular interés para nuestro grupo.
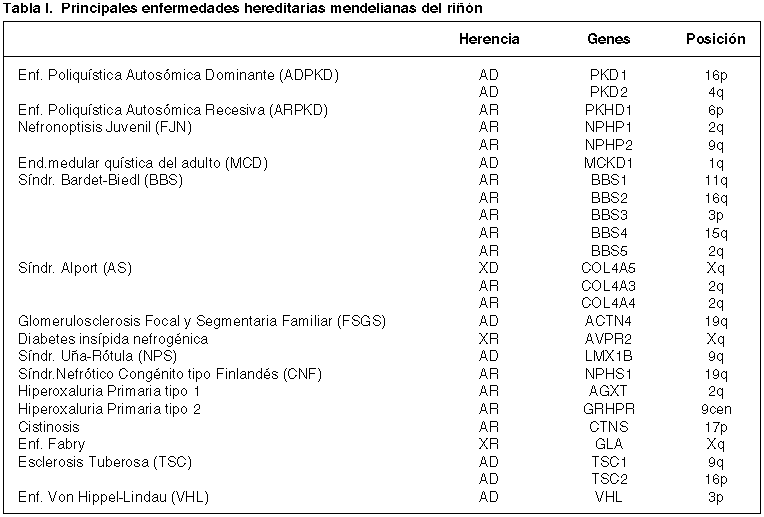
Enf. Poliquística Autosómica Dominante (ADPKD)
Esta forma de enfermedad poliquística es una de las enfermedades hereditarias mendelianas más frecuentes. Afecta a uno de cada 400-1000 individuos (3), y es responsable de alrededor del 10% de las situaciones de insuficiencia renal terminal.
En torno al 85% de los casos de ADPKD (http://www.ncbi.nlm.nih.gov/OMIM) se deben a mutaciones en el gen PKD1, localizado en 16p13.3, mientras que un 15% de los casos se deben a mutaciones en el gen PKD2, en 4q22, y casos aislados que no han podido ser asignados a ninguno de estos genes apoyan la existencia, controvertida, de otro(s) genes (PKD3...) cuya mutación podría llevar a un fenotipo similar.
El gen PKD1 es de estructura compleja (4), lo que representa retos importantes de cara al estudio molecular de mutaciones. Está constituído por 46 exones repartidos por más de 53 kb de DNA, con un mRNA de 14 kb que codifica una proteína (policistina) de 4302 amino ácidos. Esta glicoproteína de membrana parece estar implicada en interacciones célula-célula y/o célula-matriz extracelular. Contiene un extremo amino- extracelular con dominios ricos en leucina (LRR), característicos de interacción proteina-proteina, múltiples dominios transmembrana, y un extremo carboxi- con un dominio de señalización intracelular, que parece interaccionar con el producto de PKD2. Otro elemento estructural importante de la policistina es la existencia de un módulo de 80 aminoácidos, similar a dominios de la familia de las inmunoglobulinas, que se repite 16 veces. Una hipótesis probable es que PKD1 sea una molécula en la membrana basolateral que sense y vehiculice señales de interacción celular y matricial, y regule la función de PKD2, un canal iónico activado por voltaje que ejecutaría órdenes morfogenéticas (ver revisión -5-).
El patrón de expresión de la proteina es bastante amplio, no solo en túbulos renales sino tambien en conductos pancreáticos y biliares (estructuras que sufren asimismo dilataciones quísticas en esta enfermedad) e incluso en cerebro, donde los niveles son más altos. La expresión durante el periodo fetal es más llamativa que en el riñón normal de adulto, pero los riñones poliquísticos presentan niveles altos de la proteína.
Los detalles moleculares que se conocen de las proteínas PKD1 y PKD2 serían así compatibles con los factores patogénicos más conocidos sobre la formación de quistes renales: anomalías en la regulación del crecimiento-proliferación celular, en la polaridad celular, en el transporte hidroelectrolítico y en la interacción intercelular y célula-matriz.
En el diagnóstico molecular de la ADPKD hay que tener presente de entrada dos aspectos importantes:
Heterogeneidad genética: el hecho de que la enfermedad pueda estar causada por mutaciones en varios genes diferentes (PKD1, PKD2, PKD3?). Como se puede observar en la tabla I, el fenómeno de heterogeneidad genética (o de locus) es bastante frecuente y representa una de las barreras importantes a la generalización y simplificación del análisis genético molecular. Es cierto que en el caso de la ADPKD las proporciones en que el gen causal son PKD1 (85-95% de casos) es muy superior al resto, y por tanto, ante un caso nuevo es útil comenzar el estudio por PKD1. También hay evidencia de que algunos rasgos clínicos pueden ser más compatibles con mutaciones en PKD2 que en PKD1 (6), pero lo cierto es que la heterogeneidad genética complica el estudio molecular de las enfermedades hereditarias más allá de lo que podíamos prever en un principio.
Genes homólogos: los primeros 34 exones del gen PKD1 tienen muy alta homología con otras secuencias de DNA, aparentemente no funcionales (pseudogenes), ya que se transcriben en mRNA, pero no parece que se traduzcan en proteína, y han podido acumular mutaciones a lo largo de la evolución. Tres de estas secuencias homólogas están próximas a PKD1 en 16p13.1, y pueden contribuir en la génesis de mutaciones mediante el fenómeno de conversión génica (7). Consecuentemente, es muy difícil amplificar uno de estos exones con garantía que no estamos amplificando tambien secuencias de pseudogenes y no podemos estar seguros de que la posible mutacion detectada esté en el gen PKD1 o sea parte de la secuencia de un pseudogen.
Las mutaciones descritas se centran por tanto en la porción no duplicada del gen, el extremo 3’, y en esta región se han descrito cientos de mutaciones puntuales y en menor porcentaje delecciones (http://archive.uwcm.ac.uk/uwcm/mg/search/120293.html). En conjunto, parece que los datos son compatibles con que una mayoría de mutaciones tengan como consecuencia una proteína que no se localiza en la membrana o es secretada, o que en definitiva no puede llevar a cabo la señalización intracelular propia de PKD1. No se ha descrito una mutación prevalente, y parece ser que la alta frecuencia de la enfermedad en la población tiene que ver con aspectos estructurales del gen que lo hacen más susceptible a mutaciones: pseudogenes homólogos, tramos de polipurinas y polipirimidinas en los intrones 21 y 22, ...
Con estas consideraciones en mente, la estrategia práctica de diagnóstico molecular más utilizada es el análisis de ligamiento (8): un diagnóstico indirecto basado en el estudio de la segregación de marcadores polimórficos adyacentes a PKD1 en diversos miembros de la familia. Existe un marcador, conocido como KG8, que resulta muy útil en este tipo de análisis porque se localiza en el propio gen (a pocos cientos de nucleótidos del codón de parada) y es bastante polimórfico. Además de KG8, existen otros muchos marcadores polimórficos del tipo microsatélite, amplificables por PCR (tabla II), que flanquean PKD1 y son útiles en la confección de haplotipos que permitan seguir la segregación de los cromosomas en estudios de familias. Como en todo diagnóstico por ligamiento (2), existe la limitación impuesta por el tamaño de la familia, y la necesidad de contar con DNA de miembros afecto. En la práctica, los requerimientos de muestras familiares para hacer el diagnóstico molecular indirecto son: al menos 2 miembros afectos, de la misma generación, o bien 3 miembros afectos, si son de dos generaciones diferentes; y al menos 2 familiares de primer grado no afectos.

Cuando el estudio revela la falta de ligamiento a PKD1 en una familia con diagnóstico clínico de ADPKD, se investiga el posible ligamiento al gen PKD2, también mediante amplificación de microsatélites (tabla II). Ante un ligamiento positivo a PKD2, es factible amplificar cada exon del gen y hacer un rastreo de mutaciones para identificar el cambio molecular responsable de la enfermedad en esa familia (9).
Síndrome de Alport
Aunque bastante menos frecuente que la ADPKD, el síndrome de Alport es una enfermedad hereditaria relevante no sólo por su prevalencia [estimada en un caso por 50.000 nacidos vivos (10)], sino por su patrón de herencia un tanto inusual: ligado al X dominante, en la mayoría de los casos, por mutación en el gen COL4A5 en Xq22. Existe también una variante en que a la nefritis y sordera se suma leiomiomatosis esofágica difusa por mutación que afecta tanto a COL4A5 como a COL4A6, de localización contigua en el cromosoma X. En algunas familias, se observa un patrón de herencia autosómico recesivo, por mutaciones en los genes COL4A3 y/o COL4A4 (http://www.ncbi.nlm.nih.gov/OMIM). Además de esta heterogeneidad genética, hay que tener presente que hasta en un 15% de los casos se trata de mutaciones de novo, en las que la historia familiar es negativa.
La biopsia renal, y la microscopía electrónica en particular, han proporcionado claves fundamentales para el entendimiento de la enfermedad. Las irregularidades e interrupciones características de la membrana basal glomerular en los casos de Alport fueron la base de la hipótesis que implicaba a un gen de la colágena tipo 4. La triple hélice que constituye la COL4, el componente estructural más importante de la membrana basal, está formada por tres cadenas alfa seleccionadas de entre 6 posibles cadenas (A1-A6), siendo la variante más frecuente la constituída por dos A1 y una A2. Esta composición es la predominante en la matriz mesangial y la membrana basal tubular y vascular, pero la membrana basal glomerular es más rica en A3, A4 y A5, que son hélices de mayor contenido en Cys y por tanto con mayor proporción de puentes disulfuro que hacen la molécula más resistente. Esta mayor resistencia parece necesaria en estructuras como la membrana basal glomerular, que no cuenta con una red de fibras del conjuntivo subyacente.
Desde el punto de vista molecular, las secuencias de los genes codificantes de estas cadenas alfa permiten agruparlas en A1, A3, A5 por un lado, y A2, A4, A6 por otro. La distribución de todos estos genes en nuestro genoma refleja eventos de duplicación génica durante la evolución, de modo que encontramos parejas de genes adyacentes, orientados en el mismo sentido (cabeza-cola) en Xq22 (COL4A5 y COL4A6), en 2q35-37 (COL4A3 y COL4A4), y en 13q34 (COL4A1 y COL4A2). Cada gen abarca más de 100 kb, y contiene 46-52 exones que codifican proteinas de 170-185 kDa. La mayor parte de la molécula está formada por bloques de repeticiones Gly-X-Y, interrumpidas unas 21-23 veces a lo largo de este dominio colagenoso, mientras que el extremo carboxílico está integrado por un dominio no-colagenoso (NC). La posición de la Gly cada tres residuos en el dominio colagenoso es esencial para mantener la conformación en triple hélice, puesto que este pequeño aminoácido es el único que cabe en el centro de la hélice. Después de la secreción celular de triples hélices, estos monómeros se agrupan por los extremos NC para constituir dímeros y por los extremos amino pueden agruparse en tetrámeros, para constituir una malla compleja.
Se han descrito centenares de mutaciones diferentes, fundamentalmente en el gen COL4A5 (http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html), y prácticamente cada familia porta una mutación diferente. En su mayoría se trata de cambios de nucleótido que conllevan cambio de aminoácido, con frecuencia afectando a una Gly de la repetición Gly-X-Y. Las triples hélices mutadas que no se plieguen correctamente son muy susceptibles a degradación, por lo que un común denominador de muchas mutaciones diferentes es la ausencia parcial o total de hélices alfa 5, 4, y/o 3. En un 15% de los casos se han encontrado reordenaciones grandes de material genético (delecciones, inserciones, inversiones y duplicaciones), que cuando afectan también al gen COL4A6 conllevan un fenotipo con leiomiomatosis esofágica (11). Esta gran heterogeneidad alélica hace difícil el diagnóstico directo mediante análisis de DNA, por lo que generalmente se recurre al diagnóstico indirecto mediante análisis de ligamiento con marcadores microsatélites de los intrones del propio gen o adyacentes. Puesto que una mayoría de mutaciones conllevan en última instancia a la ausencia de proteína, el diagnóstico mediante anticuerpos es muy rentable en esta enfermedad. Tanto el análisis tipo Western como la inmunofluorescencia en biopsias renales o incluso de piel (12,13) permiten diagnosticar una mayoría de casos. En la interpretación de la inmunofluorescencia hay que tener en cuenta el fenómeno de inactivación de uno de los cromosomas X de la mujer, que da lugar a un patrón en mosaico, con células negativas en las que se ha inactivado la expresión del alelo sano, y otras positivas en que la inactivación aleatoria ocurrió en el cromosoma X portador de la mutación en COL4A5.
Hiperoxaluria Primaria tipo 1
A pesar de que se trata de una enfermedad muy infrecuente en general, esta causa de insuficiencia renal tiene una prevalencia significativa en nuestra comunidad. Consecuentemente, tenemos información original sobre la misma, y constituye un buen ejemplo de análisis molecular de un problema hereditario. El gen del enzima AGXT codifica una proteína de 43 kDa que se localiza en el peroxisoma, donde interviene en la detoxificación del glioxilato (http://www.ncbi.nlm.nih.gov/OMIM). Se han descrito numerosas mutaciones puntuales a lo largo de los 11 exones del gen, que se extienden en 10 kb de DNA genómico en 2q36-37. La mutación más frecuente, G170R, cuando se combina con el polimorfismo P11L, conlleva un defecto de tráfico proteico intracelular resultante en la localización mitocondrial del enzima AGXT (14) . Puesto que el glioxilato es de localización peroxisómica, la localización mitocondrial del enzima no es funcional, y los hepatocitos de estos pacientes producen abundante oxalato como resultado de la incapacidad de detoxificar el glioxilato. El acúmulo de oxalato es responsable de la nefrocalcinosis, litiasis renal, y depósitos generalizados de oxalato cálcico (oxalosis).
El diagnóstico molecular de la enfermedad se puede abordar por estudios de ligamiento, en primer lugar, estudiando la segregación de los alelos en los pedigrees familiares (fig. 2), y también de manera directa. Para el análisis directo de mutaciones en AGXT, la técnica de rastreo más útil en nuestra experiencia es la conocida como SSCP (fig. 3). Tras amplificar cada exon con primers específicos, se separan las cadenas de DNA y se someten a electroforesis en condiciones de reticulación, temperatura y glicerol optimizadas para favorecer la detección de polimorfismos conformacionales. Los exones que resultan en patrones de migración aberrantes, se someten a secuenciación directa (fig. 4). De este modo, hemos encontrado que la práctica totalidad de los alelos de hiperoxaluria primaria en nuestra comunidad llevan la mutación I244T, en combinación con el polimorfismo P11L, probablemente como consecuencia de un efecto fundacional entre los pobladores de las islas.
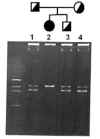
Fig. 2. Ejemplo de análisis indirecto de una enfermedad hereditaria. Esta
familia de hiperoxaluria primaria (autosómica recesiva) ha sido analizada con
un marcador polimórfico que reside en un intron del gen AGXT. Los sujetos
portadores de la mutación son heterocigotos (dos bandas, calles 1, 3 y 4),
mientras que el sujeto enfermo es homocigoto para el alelo asociado a la
mutación en AGXT (banda superior).
![]()
Fig. 3. Ejemplo de rastreo de mutaciones mediante SSCP. En este análisis
las familias de hiperoxaluria primaria se observa que el DNA amplificado de
exones, sometido a electroforesis optimizada para ver diferencias
conformacionales, permite identificar distintos patrones. Una vez secuenciados,
podemos correlacionar cada patrón con un genotipo.

Fig. 4. Autoradiograma de una reacción de secuenciación del exon 7 del gen
AGXT de un individuo homocigoto para la mutación Ile244Thr. La lectura (de
abajo hacia arriba) de las bandas de esta porción de la reacción de
secuenciación: ...AC-CTG-GAC-ACC-AAG-TGG-CTG-GCC-AAC-TTC-T... donde cada
triplete codifica... Leu-Asp-Thr-Lys-Ley-Ala-Asn-Phe-... La secuencia normal del
gen AGXT llevaría una T en lugar de la C, y codificaría el aminoácido Ile en
lugar de Thr.
ENFERMEDADES INFECCIOSAS
La detección de genomas de agentes infecciosos en biopsias renales es un procedimiento muy sensible y específico desde que contamos con la técnica de la reacción en cadena de polimerasa (PCR). La mayoría de los casos en que esta aproximación tiene utilidad en nefropatología tiene que ver con el diagnóstico de infecciones por virus: CMV, HIV, HCV, BKV, HV8...
Evidentemente, la detección de genoma viral en material de biopsia renal (algunos cortes de tejido incluído en parafina es suficiente) proporciona sólo evidencia circunstancial de que dicho virus puede jugar un papel causal en la enfermedad, pero no proporciona información sobre su nivel de actividad o contribución al deterioro funcional. No obstante, la ausencia de genoma de un determinado virus sí que sirve en muchas circunstancias para excluirlo como agente causal de la enfermedad. Otra nota de cautela que hemos de considerar es que, debido a la alta sensibilidad de la técnica, la presencia de células sanguíneas infectadas en tránsito por el tejido renal puede dar señales positivas espúreas, y evidentemente, la técnica ha de ser ejecutada con atención a la posibilidad de falsos positivos procedentes de contaminaciones en el laboratorio. Con todo, la detección molecular de agentes infecciosos se ha convertido en una de las aplicaciones con más demanda, y su uso se potencia cada vez que emergen nuevos gérmenes causantes de patología, fundamentalmente en el paciente trasplantado (herpesvirus 8, virus BK,...) (15-17). Tan pronto como contamos con secuencia parcial o total de un nuevo agente infeccioso se puede diseñar la amplificación específica de una parte de su genoma como herramienta diagnóstica, sin necesidad de esperar a que las condiciones de cultivo sean reproducibles y sensibles. Aunque tradicionalmente el diagnóstico de agentes infecciosos no ha sido parte importante de la labor en Anatomía Patológica, es cada vez más frecuente la necesidad de detectar la presencia de un germen en tejidos incluidos en parafina.
Infección por citomegalovirus (CMV)
La infección por CMV es una causa importante de morbilidad en pacientes transplantados. La detección de genoma de CMV en células de biopsias renales en pacientes infectados ha sido descrita en numerosas ocasiones, desde finales de los 80 (18). Más recientemente, el procedimiento de elección para detectar el genoma de CMV ha sido la amplificación por PCR, que permite trabajar con pequeñas cantidades de DNA purificado a partir de cortes de tejido en parafina. Este procedimiento ha mostrado una buena correlación con la detección por cultivo y serología. En nuestra Unidad de Trasplante, la monitorización de la infección por CMV se lleva a cabo mediante PCR en plasma y células sanguíneas (fig. 5). Ocasionalmente se lleva a cabo la confirmación de la infección en tejido del injerto, usando una fracción de las células obtenidas mediante citología aspirativa con aguja fina para diagnóstico de rechazo. La detección de DNA de CMV en muestras de sangre periférica (especialmente si es positivo no sólo el DNA de células sino también el DNA purificado del plasma), aunque no representa una muestra directa del injerto renal, se correlaciona bastante bien con el estado de infección por CMV, y al ser mínimamente invasiva se prefiere como procedimiento de monitorización. Con ello, el uso de la biopsia renal (cilindro o aguja fina) para diagnóstico de patología por CMV queda restringido a unos pocos casos en que se busca información adicional de tipo morfológico. En estas situaciones, la opción de la hibridación in situ, aunque más laboriosa, puede estar justificada también.
Infecciones emergentes
Ante el descubrimiento de un nuevo germen implicado en patología humana, o la descripción de gérmenes que re-emergen como patógenos importantes en determinados pacientes, surge la necesidad de un diagnóstico rápido y eficaz que no dependa de la puesta a punto de métodos tradicionales de cultivo. En los pacientes inmunosuprimidos hemos asistido en los últimos años a varias situaciones de enfermedades infecciosas emergentes o re-emergentes, y en todos ellos las herramientas moleculares han sido clave para establecer el diagnóstico preciso. La incidencia de infecciones por virus BK (15,16) y por herpesvirus 8 (17) en pacientes trasplantados renales son los ejemplos más representativos de este grupo (fig. 5). La tecnología de amplificación de DNA por PCR, una vez incorporada en un servicio diagnóstico, es fácilmente adaptable a nuevas necesidades.
![]()
Fig. 5. Detección de genoma de virus BK (panel izquierdo) y de CMV (panel
derecho) mediante PCR. El producto amplificado de 176 pb (calle 1) es el
esperado para la amplificación con los primers de BKV usados; calle 2: control
negativo, calle 3: standard. En el caso del CMV, los primers específicos usados
amplifican un fragmento de 156 pb, que es evidente en las muestras en calles 1,
2 y (más leve) 3, mientras que resulta negativo en las demás muestras; calle
10: control positivo, calle 11: standard.
NEOPLASIAS
Un aspecto en que la estrategia de clonaje posicional ha tenido más éxito en la última década ha sido definiendo el papel de genes responsables del desarrollo de neoplasias. Entre estos, la caracterización de WT1, implicado en el tumor de Wilms, y de VHL, asociado al carcinoma de células renales, son dos ejemplos importantes.
Otra contribución muy importante de la estrategia molecular al estudio de los tumores renales, se plantea en la búsqueda de perfiles de expresión génica que se asocien a la biología de la neoplasia. En los próximos años asistiremos a la subclasificación de los tumores malignos en función del perfil de expresión génica, del que probablemente se podrá precisar más el pronóstico y ayudar en el abordaje terapéutico.
Tumor de Wilms
La neoplasia abdominal más frecuente en la infancia encaja bastante bien con el modelo del doble impacto mutacional de Knudson: en las formas familiares (en torno al 1%) una copia del gen se hereda por vía germinal ya mutado, mientras que el segundo alelo sufre una mutación somática; en las formas esporádicas se requieren dos mutaciones somáticas sucesivas, que conlleven pérdida de función de ambos alelos.
Se han descrito varios loci responsables del tumor de Wilms, siendo el locus WT1 en 11p13 el que con más frecuencia se ha visto implicado en esta enfermedad. Otros loci son WT-2 en 11p15, WT-3 en 16q y WT-4 en 17q12-q21 (http://www.ncbi.nlm.nih.gov/OMIM). La asociación ocasional del tumor de Wilms a determinadas malformaciones congénitas (aniridia aislada o en conjunción con malformaciones genitourinarias y retraso mental: síndrome WAGR) ha ayudado a definir los loci implicados y a clonar los genes responsables. El gen WT1, posicionado en el mínimo intervalo de delecciones descritas en 11p13 asociadas a tumor de Wilms, se clonó y caracterizó en la última década (19,20). Se trata de un gen de 10 exones, con splicing alternativo, distribuídos en unos 50 kbp de DNA y codifica una proteína de 55 kDa con dos dominios funcionales: dominio de transactivación, implicado en la regulación de la transcripción, corresponde al extremo aminoterminal, rico en Gln y Pro; dominio de unión al DNA, corresponde al extremo carboxiterminal y contiene 4 estructuras tipo ‘dedo de Zn’ Cys-His. La proteína WT1 funciona como un factor de transcripción, que regula la expresión de genes importantes en el desarrollo del riñón y sistema genitourinario. El mecanismo a través del cual WT1 interviene en el control de la proliferación celular puede ser similar al de gen EGR1, con el que comparte homología en el dominio de unión a DNA. Posiblemente, el efecto de WT1 sea la represión de la transcripción de genes de factores de crecimiento como PDGFa, TGFb, EGFR, IGF1R, CSF1, y el propio EGR1. La importancia de WT1 en el desarrollo urogenital es subrayada por el fenotipo observado en ratones K.O. para el gen homólogo WT1 (21). Los ratones homocigóticos deficitarios en WT1 no poseen más que vestigios de yemas renales sin células del blastema, que no responden a estímulos inductores. En humanos, la delección de un alelo WT1 en línea germinal conlleva malformaciones urogenitales de variable severidad, dentro del síndrome WAGR, mientras que la pérdida funcional de ambos alelos, debida a efecto dominante negativo de una mutación puntual, heredada por línea germinal conlleva malformaciones genitourinarias severas, con esclerosis mesangial que progresa a insuficiencia renal, pseudohermafroditismo y tumor de Wilms (síndrome de Drash) (22).
Diagnóstico de mutaciones en WT1
Se han descrito casi 200 mutaciones diferentes en el gen WT1 (http://www.umd.necker.fr:2003/), distribuídas por todo el gen, aunque hay una cierta concentración en el extremo carboxi- , correspondiente al dominio de unión a DNA. En su mayoría se trata de cambios de nucleótido (muchos de ellos C>T) que conllevan cambio de aminoácido, truncación de la proteína o cambio en la pauta de lectura. También se han descrito delecciones e inserciones pequeñas, mutaciones que afectan al splicing, y ocasionales delecciones grandes del gen. Como en otros casos de genes supresores tumorales, es de esperar una pérdida funcional de ambos alelos en las células del tumor, algo que se puede determinar comparando tumor y tejido normal adyacente mediante un test de LOH (pérdida de heterocigosidad). No obstante, un porcentaje importante de mutaciones en WT1 parecen tener efecto ‘dominante negativo’, de modo que la mutación de un solo alelo conlleva la producción de una proteína anormal, capaz de asociarse con formas normales de la proteína WT1, haciendo al conjunto inactivo. Estas mutaciones, que con frecuencia se localizan en el dominio de unión a DNA, pueden verse implicadas en el desarrollo del tumor sin necesidad de que exista pérdida del alelo sano, detectable por el test de LOH.
La necesidad del diagnóstico molecular de mutaciones en WT1 se plantea muy pocas veces en la práctica, y en vista de lo comentado más arriba, los tests de LOH pueden ser útiles para determinar qué locus WT está más probablemente implicado. WT1 puede ser el gen causal a pesar de no encontrar LOH para 11p13, en cuyo caso la búsqueda de mutaciones debería empezar por los exones 7-10, del dominio de unión a DNA. Dada la abundancia de mutaciones puntuales, un rastreo de mutaciones apropiado es el basado en análisis de polimorfismos conformacionales de cadena simple (SSCP) bien a partir de exones amplificados sobre DNA genómico, o bien sobre cDNA retrotranscrito desde mRNA extraído del tumor. La secuenciación de los fragmentos que den migraciones aberrantes en SSCP suele determinar la naturaleza del cambio implicado.
Carcinoma de células renales
Las lesiones moleculares asociadas a las distintas formas de carcinoma de células renales han promovido una reclasificación de los mismos (23). La variante más frecuente, el carcinoma no papilar de células claras, tanto en su variante familiar como esporádica, se asocia a la pérdida de material genético en torno a 3p25, donde se localiza el gen VHL. En las formas familiares, siguiendo una vez más el modelo de Knudson, se hereda un alelo mutado por línea germinal, mientras que la segunda mutación ocurre en células somáticas y resulta en la pérdida funcional del gen. El síndrome de Von Hippel-Lindau (VHL) se hereda pues de manera dominante como una predisposición a una variedad de neoplasias benignas y malignas entre las que se encuentra el carcinoma de células renales. En las formas esporádicas de carcinoma no papilar de células claras, las dos mutaciones que inactivan ambos alelos del gen VHL ocurren en línea somática.
El gen VHL contiene 3 exones, con splicing alternativo del exón 2, y codifica una proteína citoplásmica de 30 kDa, que se ha visto asociada a factores de elongación de la transcripción, y por tanto podría jugar un papel en el procesado del mRNA. Las casi 800 mutaciones descritas en VHL (http://www.umd.necker.fr:2005/) se distribuyen por todo el gen, e incluyen una mayoría de cambios de nucleótido (con cambios de aminoácido, truncaciones de la proteína y aberraciones de splicing), junto a delecciones e inserciones pequeñas y delecciones grandes. Pocas veces se plantea la necesidad de diagnóstico molecular de estas mutaciones, y lo dicho para WT1 aplica a este caso también, quizá con la ventaja de que el menor número de exones hace más fácil el análisis.
PERFIL MOLECULAR DE INFLAMACIONES RENALES, INCLUYENDO PATOLOGÍA DEL TRASPLANTE
Desde los años 60, el estudio histológico de las biopsias renales ha generado un conjunto de información, predominantemente descriptiva, sobre las enfermedades renales. La relevancia de esta contribución queda reflejada en el uso de criterios morfológicos como base de clasificación de las enfermedades renales. El análisis de eventos moleculares en tejido de biopsia renal ha abierto nuevas perspectivas en el conocimiento de estas enfermedades, pero su aplicación no se ha generalizado por problemas esencialmente tecnológicos. Si comparamos el avance en la aplicación de la perspectiva molecular nefropatología con el ocurrido en otras áreas, como las neoplasias, parece claro que una limitación importante a la realización de estudios moleculares en biopsias renales es la dificultad de obtener suficiente tejido. Consideraciones éticas limitan la cantidad de tejido renal tomado en la biopsia, y por otro lado, el diagnóstico convencional en nefropatología ya demanda bastante material, de cara a estudiar tanto tejido incluido en parafina como cortes de congelación (inmunofluorescencia) y microscopía electrónica. El desarrollo de metodologías moleculares suficientemente sensibles y su integración en equipos de patólogos se presentan como una opción práctica de futuro para sacar un máximo de información, morfológica y molecular, de las muestras limitadas de tejido renal disponibles.
La sospecha de que un gen está implicado en la patogenia de una enfermedad renal requiere la detección de su mRNA o proteína. Si dicho gen se expresa normalmente en el riñón, los métodos han de ser suficientemente cuantitativos como para detectar cambios significativos en los niveles de expresión. Una limitación importante en este sentido es la dificultad en obtener muestras control válidas, puesto que nunca se realiza una biopsia renal en un sujeto sano. Aunque no sea un control ideal en muchos casos, los laboratorios en centros con programa de trasplante renal ocasionalmente tenemos acceso a riñones de cadáver que han sido descartados para trasplante. El nivel de expresión de muchos genes va a reflejar el estado del donante, el tiempo de isquemia, los procedimientos de perfusión, ..., pero a pesar de todo estos riñones representan una de las pocas fuentes de material control de que disponemos.
Los procedimientos de análisis cuantitativo mejor establecidos (Northern y ensayo de protección de RNasa) requieren demasiado material de partida, por lo que a penas tienen aplicación en nefropatología. Por otro lado, la hibridación in situ, que puede realizarse sobre el mismo material incluido en parafina para estudio morfológico, es dificilmente cuantificable. Los métodos basados en la síntesis de cDNA a partir de mRNA (RT, o transcripción reversa) y posterior amplificación del cDNA mediante PCR se presentan como la única alternativa viable cuando la fuente de material es tan escasa. De hecho, se puede llevar a cabo RT-PCR para detectar mRNAs en secciones tisulares, e incluso en regiones específicas de la nefrona, obtenidas por microdisección a partir de cortes de congelación o parafina. En los últimos años, se ha extendido el uso de RT-PCR con análisis en tiempo real, lo que ha facilitado una visión más dinámica del proceso de amplificación, y la selección de condiciones en que la cuantificación de la señal sea reproducible.
Es aún bastante limitada la cantidad de información disponible sobre los niveles de expresión de genes concretos implicados en las glomerulonefritis. Las categorías de genes de interés en este tipo de análisis incluyen los siguientes grupos:
1) Genes de respuesta endotelial.
2) Genes implicados en la presentación de antigenos, y estimulación de la respuesta inmune.
3) Genes de citoquinas y mediadores de la respuesta inmune.
4) Genes de respuesta inflamatoria (aferente), lesiva al injerto.
5) Genes de daño tisular, sea de causa inmune o no (reperfusión...).
6) Genes de reparación/remodelación tisular.
7) Genes de nefrotoxicidad por inmunosupresores.
8) Genes indicativos de expresión basal y de representación de las distintas células renales en la muestra.
Los estudios publicados hasta la fecha con tejido humano son limitados en su enfoque al análisis de la expresión de un número muy reducido de genes, utilizando RT-PCR o hibridación in situ, fundamentalmente. No obstante, la experimentación animal y trabajos en cultivo celular han potenciado un mejor entendimiento de los mecanismos básicos que median las lesiones glomerulares y del órgano trasplantado. Con el conocimiento detallado del genoma humano, se hace posible investigar el papel desarrollado por cualquier gen o familia de genes. Un avance tecnológico muy importante en este aspecto ha sido la capacidad de colocar cientos o miles de sondas moleculares en una matriz sólida de tamaño (micromatrices de DNA o DNA-microarrays). Estos diseños se están demostrando útiles en el estudio de la expresión génica de gran cantidad de genes simultáneamente, partiendo de una pequeña muestra de tejido.
Es predecible que en los próximos años podamos realizar un estudio del perfil de expresión génica de una selección de cientos de genes en una sola hibridación molecular, utilizando micromatrices de DNA, y que el análisis de la expresión relativa de estos genes ayude a definir los procesos inflamatorios renales desde un punto de vista más fisiopatológico que morfológico. La correlación entre estos perfiles de expresión y los datos morfológicos y clínicos va a ser clave en la interpretación de los mismos, por lo que se van a imponer en una primera fase los estudios multicéntricos encaminados a comprobar la reproducibilidad del método. La tecnología de las micromatrices de DNA es bastante acorde a este plan, ya que es mucho más eficaz imprimir micromatrices en un solo centro, para ser distribuídas entre los grupos participantes. En definitiva, los requisitos tecnológicos parecen haberse alcanzado recientemente, y ahora se abre un periodo de validación de algunas de estas posibilidades diagnósticas, con la esperanza que en un futuro inmediato el informe de una biopsia renal incluya no sólo datos morfológicos sino también un perfil de expresión génica. No cabe duda que esto potenciaría el valor diagnóstico, pronóstico y de orientación terapéutica de la biopsia renal.
AGRADECIMIENTOS
Nuestra investigacion esta financiada por el Ministerio de Sanidad, proyecto FIS-99/0155.
La ayuda de los Dres. Armando Torres e Ysamar Barrios, el doctorando Alfredo Santana y la tecnico Fatima Plasencia ha hecho posible esta investigacion.
BIBLIOGRAFÍA
Salido E. Análisis del DNA. I. Nefrología 1995; 15 (S2): 18-30.
Salido E. Análisis del DNA. III: Mapping genético y mapping físico. Nefrología 1995; 15 (S2): 44-57.
Gabow PA: Autosomal dominant polycystic kidney disease. New Eng J Med 1993; 329: 332-42.
European Polycystic Kidney Disease Consortium: The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 1994; 77: 881-94.
Arnaout MA: Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52: 93-123.
Parfrey PS, Bear JC, Morgan J, Cramer BC, McManamon PJ, Gault MH, Churchill DN, Singh M, Hewitt R, Somlo S, et al. The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323: 1085-90.
Watnick TJ, Gandolph MA, Weber H, Neumann HP, Germino GG. Gene conversion is a likely cause of mutation in PKD1. Hum Mol Genet. 1998; 7: 1239-43.
Coto E, Sanz de Castro S, Aguado S, Alvarez J, Arias M, Menéndez MJ, López-Larrea C. DNA microsatellite analysis of families with autosomal dominant polycystic kidney disease types 1 and 2: evaluation of clinical heterogeneity between both forms of the disease. J Med Genet 1995; 32: 442-5.
Viribay M, Hayashi T, Telleria D, Mochizuki T, Reynolds DM, Alonso R, Lens XM, Moreno F, Harris PC, Somlo S, San Millan JL. Novel stop and frameshifting mutations in the autosomal dominant polycystic kidney disease 2 (PKD2) gene. Hum Genet 1997; 101: 229-34.
Pajari H, Kaariainen H, Muhonen T, Koskimies O. Alport’s syndrome in 78 patients. Epidemiological and clinical study. Acta Paediatr 1996; 85: 1300-6.
Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem. 1993; 268: 26033-6.
Lajoie G. Approach to the diagnosis of thin basement membrane nephropathy in females with the use of antibodies to type IV collagen. Arch Pathol Lab Med 2001; 25: 631-6.
van der Loop FT, Monnens LA, Schroder CH, Lemmink HH, Breuning MH, Timmer ED, Smeets HJ. Identification of COL4A5 defects in Alport’s syndrome by immunohistochemistry of skin. Kidney Int 1999; 55: 1217-24.
Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation. Proc Nat Acad Sci 1991; 88: 10900-4.
Petrogiannis-Haliotis T, Sakoulas G, Kirby J, Koralnik IJ, Dvorak AM, Monahan-Earley R, De Girolami PC, De Girolami U, Upton M, Major EO, et al. BK-Related Polyomavirus Vasculopathy in a Renal-Transplant Recipient. N Engl J Med 2001; 345: 1250-5.
Rosen S, Harmon W, Krensky AM, Edelson PJ, Padgett BL, Grinnell BW, Rubino MJ, Walker DL. Tubulo-interstitial nephritis associated with polyomavirus (BK type) infection. N Engl J Med 1983; 308: 1192-6.
Diociaiuti A, Nanni G, Cattani P, Lesnoni La Parola I, Masini C, Capuano M, Pozzetto U, Fadda G, Castagneto M, Cerimele D. HHV8 in renal transplant recipients. Transpl Int. 2000; 13 (S1): S410-2.
Gnann JW Jr, Ahlmen J, Svalander C, Olding L, Oldstone MB, Nelson JA. Inflammatory cells in transplanted kidneys are infected by human cytomegalovirus. Am J Pathol 1988; 132: 239-48.
Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990; 60: 509-20.
Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990; 343: 774-8.
Mueller RF. The Denys-Drash syndrome. J Med Genet1994; 31: 471-7.
Storkel S, Eble JN, Adlakha K, Amin M, Blute ML, Bostwick DG, Darson M, Delahunt B, Iczkowski K. Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 1997; 80: 987-9.
![]()