
Vol. 35, n.º 2, 2002
|
REVISTA
ESPAÑOLA DE
Vol. 35, n.º 2, 2002 |
José María Ezpeleta1, Alfredo López Cousillas2
1 Servicio de Obstetricia y Ginecología. 2 Servicio de Anatomía Patológica. Hospital Universitario Virgen del Camino, Pamplona.
RESUMEN
La enfermedad trofoblástica gestacional agrupa a diferentes entidades interrelacionadas: mola completa, generalmente diploide con origen cromosómico paterno, mola parcial generalmente triploide, tumor trofoblástico del lecho placentario y coriocarcinoma, con tendencias variables a la invasión local y a las metástasis, cuyo denominador común es la hipersecreción de hCG. El coriocarcinoma es diploide y proviene de ambos progenitores, excluyendo probablemente su origen directo en la mola completa. El tumor trofoblástico del lecho placentario está constituido por trofoblasto mononuclear intermedio no conteniendo vellosidades coriónicas e inmunohistoquimicamente caracterizado por expresar muchas de sus células hPL y unas pocas hCG. En el artículo, desarrollamos una revisión y actualización de aspectos clínicos y morfológicos de la enfermedad trofoblástica gestacional.
Palabras clave: Enfermedad trofoblástica gestacional. Tumores trofoblásticos gestacionales.
SUMMARY
Gestational trophoblastic diseases includes a group of interrelated entities: complete mole, generally diploid and from paternal chromosomes origin, partial mole generally triploid, placental-site trophoblastic tumor, and choriocarcinoma, that have varying propensities for local invasion and metastasis, with a common denominator which is a hypersecretion of hCG. The choriocarcinoma is diploid and its genetics material comes from both parents, this probably excludes a direct filiation between complete mole and choriocarcinoma. The placental-site trophoblastic tumor is composed almost entirely of mononuclear intermediate trophoblast and does not contain chorionic villi, and immunohistochemical staining reveals many hPL-producing cells, and few hCG-producing ones. This report will review and update main clinical and morphologic data of the gestational trophoblastic diseases.
Key words: Gestational trophoblastic diseases. Gestational trophoblastic tumors.
Se conoce con el nombre de enfermedad trofoblástica gestacional (ETG) a un conjunto de procesos benignos y malignos poco habituales, derivados de una proliferación anormal del trofoblasto de la placenta humana (hiperplasia) y del genoma paterno, con una contribución materna ocasional; incluye la mola hidatiforme completa invasiva o no, la mola parcial y los tumores trofoblásticos gestacionales, coriocarcinoma y tumor del lecho o sitio placentario (TSP) (1). Es extremadamente raro hallar trastornos similares en otras especies diferentes a la humana. Estas enfermedades se consideran peculiares, ya que el producto de la concepción a partir del cual se originan, es genéticamente extraño al anfitrión materno.
La ETG representa un espectro único de patologías interrelacionadas con el denominador común de una hipersecrección de gonadotropina coriónica (hCG), que constituye un marcador tumoral sensible que se correlaciona bien con la progresión y persistencia de la enfermedad, excepto el TSP que produce lactógeno placentario (hPL) (2), con tendencias variables a la invasión local y a las metástasis y se encuentra entre las raras enfermedades que se pueden curar, incluso en casos de extensa diseminación (3).
La mola hidatiforme es un producto de la concepción que se caracteriza por una hiperplasia trofoblástica y por la tumefacción edematosa de las vellosidades coriónicas, cuyo diámetro oscila entre 0,1 y 3 cm, adquiriendo la morfología de bandas y cúmulos de vesículas, que confieren el típico aspecto de «racimos de uvas».
La variedad de mola hidatiforme más fácilmente identificable es la completa o clásica, en la que falta el feto y donde todas las vellosidades presentan degeneración hidrópica (fig. 1) y son avasculares aunque puede observarse algún vaso degenerado, siendo notable la hiperplasia del citotroblasto y del sincitiotrofoblasto (4). Se suele identificar precozmente por un patrón ecográfico característico, descrito hace muchos años (5).

Fig. 1. La
principal característica de la mola completa es la degeneración hidrópica de
la mayoría de las vellosidades y la proliferación del trofoblasto, que puede
ser llamativa o mínima según cada caso (H.E., Neg. X40).
El segundo tipo de mola hidatiforme, la parcial, presenta al mismo tiempo características de una placenta de desarrollo normal y de una mola hidatiforme completa, con una gama de vellosidades desde normales a quísticas (fig. 2), mientras que la hiperplasia trofoblástica es sólo focal y por lo general afecta a la capa sincitiotrofoblástica y se asocia con el festoneado del contorno de las vellosidades (fig. 3) y la presencia de inclusiones trofoblásticas en el estroma (6). En algunos casos de mola hidatiforme parcial está presente el feto, pero su desarrollo es casi siempre anormal, y aunque en la mayoría de los casos el feto no está presente, puede deducirse la existencia de desarrollo fetal por la demostración de hematíes nucleados en los vasos sanguíneos de las vellosidades, es decir fetales (7).

Fig. 2. La
mola parcial tiene dos poblaciones de vellosidades coriales: unas son de tamaño
normal y otras son marcadamente hidrópicas (H.e., Neg. X40).

Fig. 3. En
la mola parcial son frecuentes las vellosidades con contornos festoneados y al
menos algunas vellosidades contienen cisternas centrales (H.E., Neg. X40).
El carácter propio de la mola hidatiforme parcial se confirmó por los estudios sobre la constitución genética, generalmente triploide, o aneuploide con trisomía para un cromosoma, sugiriendo que las molas hidatiformes comprendían dos entidades diferentes no solo anatomopatológicamente, sino también desde un punto de vista genético (8,9). Dicho estudio, definió las molas hidatiformes parciales, como aquellos productos de las concepción en los que existía un feto, cordón o membranas amnióticas con vellosidades normales e hidrópicas; la hiperplasia, presente en algunos casos, no era importante según ellos. En actualidad se acepta que la hiperplasia es un elemento esencial para establecer el diagnóstico anatomopatológico de mola hidatiforme (10).
Los autores ya mencionados, pudieron comprobar que las molas hidatiformes completas eran diploides y las parciales, generalmente triploides (8,9). La definición anatomopatológica y genética de estas dos entidades de mola hidatiforme fue confirmada más tarde por otros autores (11,12) (tabla I).

Los tumores trofoblásticos gestacionales, también llamados ETG persistente (ETGP), ocurren tras la evacuación o el fin de la gestación y pueden proceder, no sólo de la mola hidatiforme, sino después de cualquier suceso gestacional, incluido abortos, terapéuticos o espontáneos, gestaciones ectópicas y embarazos a término.
El término tumor trofoblástico gestacional, comprende la mola invasiva y los tumores malignos coriocarcinoma y tumor trofoblástico del lecho o sitio placentario e incluye a procesos con secreción anormalmente elevada y prolongada de hCG más de 6 semanas después del suceso gestacional. Esta secreción es independiente de la naturaleza del trofoblasto, que puede ser malformativo y de crecimiento limitado en las molas parciales y completas o neoplásico, de crecimiento ilimitado y rápido en el coriocarcinoma o lento en el TSP.
En la mola invasiva se produce la invasión del miometrio o de sus vasos sanguíneos por células del citotrofoblasto y del sincitiotrofoblasto; este proceso se distingue morfológicamente por las persistencia de las vellosidades placentarias, que no se observan en el coriocarcinoma ni en el TSP.
El coriocarcinoma en la proliferación atípica del trofoblasto (fig. 4), comprende tanto al citotrofoblasto como al sincitiotrofoblasto, sin presencia de estroma; es típico que la lesión esté formada por un área central de tejido necrótico, rodeada por un anillo periférico de tejido tumoral (13). No se puede deducir la clase de gestación previa a partir de las características histopatológicas; sin embargo la naturaleza de dicha gestación previa es importante en la consideración de la genética del coriocarcinoma.

Fig. 4. El
coriocarcinoma es un tumor maligno compuesto de trofoblasto y que carece de
vellosidades coriales (H.E., Neg. X100).
El tumor trofoblástico del lecho placentario fue descrito en 1976 como un pseudotumor no neoplásico, pero posteriormente se definió anatomopatológicamente como entidad propia (14), y se determinó su potencial maligno, al observarse que algunas pacientes desarrollaban metástasis y fallecían por la enfermedad (15); se han descrito unos 100 casos en el mundo y la mortalidad estimada oscila entre un 15 y un 20% (16). En 1995 se describieron dos casos en España, en mujeres de 25 y 27 años que manifestaron la enfermedad con hemorragia genital persistente, con antecedentes en ambos casos de abortos previos 3 años antes (17). Se ha descrito la eritrocitosis como patología asociada al tumor, resolviéndose la policitemia tras la histerectomía (18).
Recientemente se ha publicado el caso de una paciente con prolongada remisión (19) tras metástasis consecuentes a tumor del lecho placentario tratada con quimioterapia. También recientemente se ha expuesto la experiencia de 7 casos de TSP, 3 de los cuales fueron tratados con histerectomía por tumor confinado al útero, 2 evolucionan satisfactoriamente y continúan con quimioterapia por sus metástasis pulmonares y 2 han fallecido por enfermedad metastásica a pesar de la terapia (20). El TSP es de lenta progresión.
El TSP se origina a partir del trofoblasto del lugar de implantación de la placenta y está constituido por trofoblasto intermedio (21), principalmente por citotrofoblasto mononuclear, con muy escasas células sincitiotrofoblásticas plurinucleadas; la casi total ausencia de sincitiotrofoblasto caracteriza a dicho tumor y lo diferencia del coriocarcinoma. Tiene tendencia a formar nódulos generalmente múltiples, e infiltra la decidua, el miometrio y las arteriolas espirales. La inmunohistoquímica demuestra gran cantidad de células productoras de hPL y unas pocas células productoras de hCG (22).
Desde el punto de vista genético puede proceder tanto de un producto de la concepción normal, como de una mola hidatiforme o aborto (23).
La enfermedad trofoblástica gestacional persistente, puede metastatizar más allá del cuerpo uterino, originando el estadio de la enfermedad denominado ETG metastásica o tumor trofoblástico metastásico (10). Es conveniente recordar que en el embarazo normal se produce, a veces, cierto grado de embolia trofoblástica a los pulmones, aunque es excepcional el transporte de vellosidades completas (3) (fig. 5).
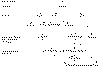
Fig. 5. Evolución
de los productos de la concepción en la ETG.
El diagnóstico de la ETG es esencialmente anatomopatológico, aunque en ciertos casos es apoyado por técnicas citogenéticas, por la citometría de flujo y la utilización de sondas moleculares.
Desde el punto de vista clínico, la ETG agrupa entidades malignas como el coriocarcinoma y el tumor del lecho placentario y otras, cuya característica clínica es el riesgo de complicaciones hemorrágicas, como es el caso de algunas molas completas (invasivas), o las raras hiperplasias trofoblásticas que complican a veces las molas parciales; otras son benignas como la mayoría de las molas parciales y las molas completas. La hemorragia genital es el síntoma más frecuente en las pacientes con mola completa, y se debe a la separación del tejido molar de la pared uterina. En general, el cuadro clínico inicial, tiene síntomas y signos de la amenaza de aborto y en ocasiones, el diagnóstico sólo se realiza tras el estudio histológico del material de un aborto, y aunque el diagnóstico, hoy en día, es mucho más precoz, el riesgo de desarrollo de enfermedad trofoblástica persistente permanece invariable.
Otros signos o patologías asociados son: la hiperemesis gravídica, de baja frecuencia por el diagnóstico precoz de la enfermedad, la preeclampsia, que se debe sospechar cuando es de aparición muy precoz en las molas parciales, el hipertiroidismo asociado, que es muy poco frecuente pero que si se sospecha debe ser tratado previamente a la evacuación de la gestación molar, ya que se puede desencadenar una crisis tiroidea, y la insuficiencia respiratoria aguda, de aparición esporádica pero grave, consecuente con la embolización trofoblástica de los vasos pulmonares.
En la exploración se puede encontrar: aumento del tamaño uterino causado tanto por el volumen del tejido coriónico, como por la existencia de coágulos retenidos dentro de la cavidad, y se correlaciona bien con los niveles de hCG; la tercera parte de las pacientes con mola completa asocian masa anexial, a veces superior a 6 cm de diámetro, son los quistes tecaluteínicos, generalmente bilaterales y multiloculares que se atribuyen a altos niveles de hCG (superiores a 100.000 mUI/ml) y suelen desaparecer espontáneamente tras la evacuación de la mola, a los 2-3 meses, pudiendo requerir tratamiento si se asocian con hemorragia y/o dolor agudo por rotura o torsión.
Las exploraciones complementarias se basan en la ecografía y en los niveles de hCG, siendo desde el punto de vista clínico más útil la subunidad b (la subunidad a es similar a las subunidades a de la LH, FSH y TSH). En la gestación normal el pico de hCG ocurre entre la semana 9 y la 14 de la gestación, por lo que una persistencia más prolongada sugiere una gestación molar.
La mola parcial, que representa entre un 10-20% de las molas hidatiformes, suele manifestarse en la clínica más tardíamente que la mola completa, y lo hace a finales del primer trimestre o a lo largo del segundo, como una amenaza de aborto. El útero suele corresponder o ser menor que el tiempo de amenorrea, siendo similares pero menos frecuentes los síntomas asociados. Casi todas se presentan con un embrión muerto al final del primer trimestre. La situación de feto vivo con ecografía sugestiva de mola, es compleja por las alteraciones cromosómicas de estos fetos; se debe realizar el diagnóstico diferencial con el aborto, el embarazo ectópico, la gestación múltiple y el hidramnios; el diagnóstico anatomopatológico del legrado es definitivo.
La primera descripción «exacta» de mola hidatiforme es del médico español Cristóbal de Vega en el año 1564 (24).
En 1839 Cruveilhier describe la mola parcial y Saenger en 1889 hace la primera clasificación de conjunto, distinguiendo tres tipos de neoplasias: sarcoma decidual o deciduocelular sin participación visible de los elementos coriales, sarcoma deciduocelular con participación de elementos coriales, y formas intersticiales destructivas, cada una de ellas con sus correspondientes subclasificaciones.
Marchand en 1895 impulsa y establece tres grupos de enfermedades trofoblásticas, afirmando taxativamente que la mola no es un tumor: mola vesicular o hidatiforme, mola destruens y corioepitelioma.
Hertig y cols. describen en 1956 un continuo evolutivo, comenzando por la mola parcial, pasando a mola completa y a mola invasiva y terminando en coriocarcinoma (25).
En 1977 Kajii y cols. determinaron el origen paterno de la mola completa (26).
Se han realizado múltiples clasificaciones de las enfermedades trofoblásticas. En 1980 Surwit y Hammond propusieron desde el punto de vista clínico, pronóstico y terapéutico la de Enfermedad Trofoblástica:
De evolución benigna:
– mola hidatídica.
De evolución maligna:
– mola invasora o corioadenoma destruens.
– coriocarcinoma* no metastásica
* metastásica- de buen pronóstico
- de mal pronóstico
En 1979 la Sociedad Internacional para el estudio de las Neoplasias Trofoblásticas, propuso la clasificación en estadios clínicos, aceptada por la OMS, la FIGO y la SEGO y diferenció:
Mola o embarazo molar, con bajo o alto riesgo
Enfermedad trofoblástica persistente (ETGP) con los estadios I-IV dependiendo de la extensión de la lesión y de que sea: con o sin metástasis y de alto o bajo riesgo
La clasificación de la ETG preconizada por la OMS (10) y que es ampliamente aceptada, se basa fundamentalmente en criterios macroscópicos tradicionales: vellosidades hidrópicas con o sin embrión y tumor sin vellosidades y lesiones diversas.
Mola hidatiforme completa o parcial.
Mola hidatiforme invasiva.
Coriocarcinoma.
Tumor trofoblástico del lecho placentario.
Lesiones trofoblásticas diversas.
– Reacción exagerada del lecho placentario.
– Nódulos y placas del lecho placentario (27).
Lesiones trofoblásticas no clasificadas.
También se acepta desde el punto de vista anatomopatológico la clasificación (1):
Tumores que forman vellosidades: Mola parcial o completa.
Tumores sin vellosidades: endometritis sincitial, tumores del sitio placentario y coriocarcinomas.
La utilización del cariotipo, la citometría de flujo y la biología molecular, permiten también una clasificación citogenética (28):
Con origen en trofoblasto perivellositario
– Hiperplasia trofoblástica con triploidía, tetraploidía o aneuploidía.
– Mola hidatiforme completa (invasiva o no).
– Coriocarcinoma gestacional.
Con origen en el lecho placentario.
– Carcinoma trofoblástico del lecho placentario.
Básicamente la ETG se ha dividido en mola hidatiforme y en tumores trofoblásticos gestacionales (29).
Clasificación de Enfermedad Trofoblástica Gestacional según el Colegio Americano de Obs tetras y Ginecólogos:
Mola Hidatiforme
– Completa
– Parcial
Tumores Trofoblásticos Gestacionales
– No metastásicos
– Metastásicos* Bajo riesgo sin factores de riesgo.
* Alto riesgo algún factor de riesgo.- Niveles de hCG previos al tratamiento > 40000 mU/mL
- Duración > 4 meses
- Metástasis en hígado o cerebro
- Fracaso de tratamiento quimiterápico previo
- Antecedente de embarazo a término.
El término de neoplasia trofoblástica gestacional no es muy empleado, porque la mola invasiva no supone la formación de tejido nuevo de carácter tumoral (30).
Desde el punto de vista clínico es importante el estadiaje de los tumores trofoblásticos gestacionales propuesta por la Federación Internacional de Ginecología y Obstetricia (FIGO):
Estadio I. Enfermedad confinada al útero.
– Ia. sin factores de riesgo
– Ib. con un factor de riesgo.
– Ic. con dos factores de riesgo.
Estadio II. Tumor trofoblástico gestacional que se extiende fuera del útero, pero que se limita a los elementos genitales (anejos, vagina, ligamentos anchos).
– IIa. sin factores de riesgo.
– IIb. con un factor de riesgo.
– IIc. con dos factores de riesgo.
Estadio III. Enfermedad trofoblástica gestacional que se extiende hasta los pulmones, con o sin afectación de vías genitales conocida.
– IIIa. sin factores de riesgo.
– IIIb. con un factor de riesgo.
– IIIc. con dos factores de riesgo.
Estadio IV. Todos los otros sitios de metástasis.
– IVa. sin factores de riesgo.
– IVb. con un factor de riesgo.
– IVc. con dos factores de riesgo.
Los factores de riesgo que afectan al estadiaje son:
HCG >100.000 mU/ml y duración de la enfermedad mayor de 6 meses a partir de la terminación del embarazo precedente. Deben considerarse los siguientes factores de seguimiento y pronóstico y anotarse en el historial clínico en caso positivo: se administró quimioterapia previa por tumor trofoblástico gestacional diagnosticado; deben informarse por separado los tumores del sitio placentario; no se requiere verificación histológica de la enfermedad.
El estadiaje permite la comparación objetiva de los datos estadísticos sobre resultados aportados por los distintos autores en sus publicaciones.
Estadio I. Pacientes con concentraciones de hCG persistentemente elevadas y tumor confinado al cuerpo uterino
Estadio II. Pacientes con metástasis hacia vagina, pelvis o ambas.
Estadio III. Pacientes con metástasis pulmonares con afección uterina, vaginal o pélvica o sin ella. (El diagnóstico se basa en la concentración creciente de la hCG en presencia de lesiones pulmonares en las radiografías u otras técnicas de imagen).
Estadio IV. Pacientes con enfermedad avanzada y afección de cerebro, hígado, riñón o tubo digestivo. Estas pacientes se encuentran en la categoría de peor riesgo, porque en ellas el tumor será casi seguramente resistente a la quimioterapia. Suele encontrarse el patrón histológico del coriocarcinoma, y a menudo la enfermedad surge después de un embarazo no molar.
Además de la clasificación basada en el estadiaje, es importante considerar variables para pronosticar la probabilidad de resistencia farmacológica y ayudar a seleccionar la quimioterapia apropiada. La OMS ha propuesto un sistema de puntuación pronóstica que permite predecir, con toda confianza el potencial de que ocurra resistencia a la quimioterapia. Las pacientes con enfermedad en estadio I suelen tener una puntuación de bajo riesgo y lógicamente las pacientes en estadio IV tienen una puntuación de alto riesgo, por lo que se aplica fundamentalmente a pacientes en estadios II y III.
Este tipo de clasificaciones por etapas posibilita protocolizar el tratamiento de los tumores trofoblásticos gestacionales persistentes.
Las destacadas posibilidades curativas de la ETG, incluso en presencia de metástasis amplias pueden ser parcialmente atribuidas a la respuesta inmunológica, de la madre portadora, a los antígenos paternos expresados en las células trofoblásticas (3). El pronóstico de pacientes con coriocarcinoma gestacional es mejor si se evidencia la infiltración de linfocitos y monocitos en el límite entre el tumor y el tejido materno (31), porque los linfocitos y macrófagos que infiltran el coriocarcinoma gestacional son probablemente expresión de las oncoproteínas y antígenos paternos, muy activadores de las células inmunitarias.
La ETG constituye una situación única de tumor humano naturalmente injertado parásito de la madre. Como aloinjerto el tejido trofoblástico con constitución antigénica diferente a la madre, puede ser rechazado espontáneamente, pero también se adapta, prolifera y constituye un tumor invasivo. Las células inmunológicas activas promueven la regresión de los tumores trofoblásticos gestacionales a través de la liberación de citocinas. Está bien documentado que las citocinas inhiben la proliferación de células de coriocarcinoma, in vitro y del incremento de la expresión de HLA en las mismas células, también in vitro, incrementando de esa manera la inmunogenicidad (32).
Se ha preconizado que el desarrollo y progresión de la ETG persistente podría estar favorecida por la histocompatibilidad de la paciente y su pareja. Si la paciente y su pareja son histocompatibles, el tumor trofoblástico que contiene antígenos paternos puede no ser inmunogénico para la madre portadora. La intensidad de la respuesta inmunológica en la portadora se relaciona con la inmunogenicidad del tumor trofoblástico. Sin embargo la histocompatibilidad entre la paciente y su pareja no es un requisito previo para el desarrollo y progresión de la ETG persistente. Por otra parte el sistema HLA influye en la evolución progresiva y fatal de los tumores trofoblásticos gestacionales. Se ha comunicado (33) que la resistencia a la quimioterapia en coriocarcinoma se asocia a incrementos en la histocompatibilidad entre la paciente y su pareja. De forma similar se ha observado que la histocompatibilidad entre la paciente y su pareja va asociada a un grave riesgo de metástasis en tumores trofoblásticos (34). La mola completa tiene cromosomas sólo de origen paterno, induciendo una intensa respuesta inmunológica de la madre. Comparado con placentas normales, la mola completa tiene cinco veces más infiltración de células T (35). En los inmunocomplejos circulantes, en pacientes con mola completa, ha sido establecido el contenido de antígeno HLA paterno (36). La madre con mola completa está por tanto, sensibilizada a los antígenos paternos HLA A, B, C. La distribución de antígenos HLA en las vellosidades coriónicas de la mola ha sido determinada por inmunofluorescencia. Los antígenos HLA A, B, C, han sido detectados en las células del estroma de las vellosidades coriónicas de la mola, pero no en el trofoblasto velloso.
La madre puede ser sensibilizada por los antígenos paternos HLA, cuando el trofoblasto velloso es alterado, y células del estroma velloso HLA positivas son liberadas en la circulación materna.
Algunas mujeres con cierto desequilibrio genómico podrían tener una predisposición constitucional para la ETG. Se han descrito casos de enfermedad trofoblástica en mujeres con mosaico trisómico (37).
Las técnicas de estudio utilizadas son múltiples (38). El análisis citogenético en metafase permite estudiar la ploidía del ADN por citometría de flujo, y el origen genético por análisis de los polimorfismos cromosómicos y enzimáticos y de la longitud de los fragmentos de restricción en molas y tejidos de los progenitores con PCR.
La citometría de flujo detecta variaciones de ploidía entre el 4 y el 10%, variaciones más débiles no son detectadas por este método. La hibridación «in situ» en interfase explora igualmente la ploidía y el sexo (39). La utilización de oncogenes, c-myc, c-ras, c-erb-B2, bcl-2 (40,41), podría ayudar a precisar el pronóstico de los tumores trofoblásticos, pero no se ha encontrado correlación con el riesgo de persistencia de una enfermedad trofoblástica gestacional (42,43), y actualmente no existe ningún marcador genético para predecir la evolución y potencialidad de la ETG (33). Incrementos en la expresión de p53 y c-fms han sido observados en molas completas (44,45), e incrementos de ras y c-myc ARN han sido cuantificados en coriocarcinomas (46).
Recientemente se ha investigado la expresión de varios factores de crecimiento y oncogenes en la placenta normal, en la mola completa y parcial y en el coriocarcinoma (47). La mola completa y el coriocarcinoma están caracterizados por la sobreexpresión de c-myc, c-erb B2 y bcl-2, y estas oncoproteínas son importantes en la patogénesis de la ETG. La expresión de la proteína c-fms no se diferencia entre la placenta normal y la enfermedad trofoblástica. La mola completa y el coriocarcinoma se caracterizan además, por incrementos de expresión de p53, p21, Rb y MDM2 (46).
Durante los acontecimientos epigenéticos, ciertos genes se expresan normalmente en cualquiera de los alelos maternos o paternos. Alteraciones en el encuentro entre los alelos maternos y paternos, en la metafase de la primera división meiótica del cigoto han sido asociadas con molas completas y coriocarcinomas (48,49), jugando un importante papel en la patogénesis de los tumores trofoblásticos gestacionales. El descubrimiento del origen paterno de la mola hidatiforme completa, gracias a la utilización del polimorfismo en banda, data de 1977 (26). Generalmente las molas completas son diploides, comprendiendo dos juegos de cromosomas paternos con la persistencia de un ADN mitocondrial materno. En el 85-90% de los casos el cariotipo es 46XX y en el 10% 46 XY (50). Analizando el origen genético y las técnicas de biología molecular, entre el 60 y 70% de las molas completas son homocigotas y resultado de la fertilización de un óvulo vacío, sin núcleo o inactivado (51), por un espermatozoide haploide con duplicación del genoma masculino (espermio 23 X que se duplica a 46 XX) (52), el concepto 46 XY es teóricamente posible pero no viable, el cariotipo XY es siempre letal, resultando un cigoto diploide pero con sólo ADN genéticamente paterno y ADN mitocondrial materno. El resto de las molas completas son heterocigotas y resultantes de la fertilización de un óvulo anucleado o inactivado por dos espermatozoides con lo que el cigoto diploide resulta 46 XY o 46 XX, pero siempre de origen enteramente paterno (53) (fig. 6).

Fig. 6. Mecanismo
de formación de molas completas diploides.
Existe un tercer mecanismo descrito en las molas completas homocigotas, resultado de errores en la segunda división meiótica del espermatozoide (26), con el resultado de un espermatozoide diploide, pero que no se ha encontrado nunca, y aunque está demostrada la existencia en sujetos sanos de espermatozoides diploides 46XX, 46XY, 46YY, su participación en la génesis de molas completas está por demostrar.
Algunas raras molas completas son aneuploides (haploides o tetraploides). Se ha demostrado también que el 20% de las molas completas son de origen mixto (39). Estas molas completas se encuentran en gestaciones múltiples asociando un desarrollo embrionario normal y una mola completa, con serias y complejas implicaciones clínicas, ya que los tumores trofoblásticos gestacionales ocurren comúnmente tras mola completa (3). Aproximadamente el 60% de mujeres con embarazo gemelar, coexistiendo feto evolutivo normal y mola completa, requieren quimioterapia por tumor trofoblástico gestacional persistente (54). Se ha descrito que el 70% de los casos requieren evacuación, antes de la viabilidad fetal, con serias complicaciones hemorrágicas o preeclampsia muy grave (55).
La mayoría de las molas parciales son triploides, mola embrionada, diferenciándose de las molas completas en que retienen parte de la contribución genética materna. Contienen 69 cromosomas (XXY el más habitual, XYY, XXX) el cariotipo YYY no se ha encontrado nunca ya que es letal. Algunas y raras molas parciales son aneuploides (haploides o tetraploides). La frecuencia de aparición de mola parcial en los productos abortivos (de aquí la trascendencia del estudio de la unidad feto-placentaria (56)), oscila entre en 10 y un 15% (57). El 85 % de las triploidías corresponde a un fenotipo masculino, un ovocito haploide es fecundado por dos espermatozoides haploides. El 15 % de las molas parciales corresponden a un fenotipo femenino (58), resultando de la fecundación de un ovocito diploide por un espermatozoide haploide. La existencia de dos juegos de cromosomas maternos favorece el desarrollo de un feto anormal asociado a un trofoblasto sin aspecto molar. Por el contrario, la existencia de dos juegos cromosómicos de origen paterno, da como resultado una placenta de aspecto molar y el desarrollo de un feto normal (57). De todos modos, esta distinción de fenotipos no es absoluta. Aunque la mola parcial pocas veces origina enfermedad trofoblástica persistente metastásica (sólo el 2,3% frente al 13-20% en las molas completas), y la mayoría de las que la originan son triploides, los casos descritos con mola parcial diploide son menos sensibles a la quimioterapia, aunque otros autores no encuentran relación con la ploidía (57).
El concepto de impresión genómica es importante para comprender el desarrollo de la ETG. Las diferencias biológicas entre las distintas categorías de molas hidatiformes dependen del genoma nuclear. La presencia de dos juegos de cromosomas paternos en una mola hidatiforme completa diploide origina un fallo en el desarrollo embrionario y favorece la proliferación del trofoblasto. En la mola hidatiforme parcial triploide la presencia de un juego de cromosomas maternos, además de dos juegos paternos, es compatible con el inicio del desarrollo fetal, pero también con más proliferación trofoblástica que en la placenta normal (59). El desarrollo de un coriocarcinoma es considerablemente más frecuente después de una mola hidatiforme que de un embarazo normal, lo cual sugiere que la presencia de dos juegos de cromosomas paternos puede constituir un factor de riesgo; en cambio raras veces aparecen secuelas malignas después de una mola hidatiforme parcial, lo cual sugiere a su vez que la presencia de un juego de cromosomas maternos puede constituir un factor contrario a la persistencia de la ETG. El factor pronóstico asociado a la composición genética es difícil de comprender. Globalmente la progresión hacia la ETG persistente es del 20%, admitiéndose que el riesgo se acrecienta en las molas heterocigotas, y más especialmente en las molas con cariotipo XY, no mostrando el análisis genético ninguna diferencia en la tasa de resistencias a la quimioterapia ni en el número de ETG metastásica (60). En la práctica, la biología molecular precisa el origen parental de los tejidos en la ETG, no pudiendo prejuzgar su evolución, independiente de que sean benignas (molas parciales), presumiblemente benignas (molas completas) o malignas (tumores trofoblásticos gestacionales).
BIBLIOGRAFÍA
Mazur MT, Kurman RJ. Gestational trophoblastic disease and related lesions. En: Kurman RJ editor. Blaunstein’s pathology of the female genital tract. 4th ed. New York: Springer-Verlag. 1994, p. 1049-93.
Kurman RJ, Young RH, Norris HJ, Main CS, Lawrence WD, Scully RE. Immunocytochemical localization of placental lactogen and chorionic gonadotrophin in the normal placenta and trophoblastic tumors, with emphasis on intermediate trophoblast and the placental site trophoblastic tumor. Int J Gynecol Pathol 1984; 3: 101-21.
Berkowitz RS, Golstein DP. The management of molar pregnancy and gestational trophoblastic tumours. En Knapp RC, Berkowitz RS, editores. Gynecologic Oncology, 2nd ed. New York: Mc Graw-Hill 1992, p. 328-38.
De Agustín P, Ruiz A, López F, Contreras F. Patología de la enfermedad trofoblástica. Simposio Enfermedad Trofoblástica 1972; 79-98.
Salem S. Ultrasound diagnosis of trophoblastic disease. En: Sanders RC, James AE(Jr) editores. Ultrasonography in Obstetrics and Gynaecology. New York: Appleton-Century Crofts: 1977; p. 255-66.
Silverberg SG, Kurman RJ. Tumors of the uterine corpus and gestational trophoblastic disease. En: Rosai J, Sobin LJ, editores. Atlas of tumor pathology: tumors of the uterine corpus and gestational trophoblastic disease, fasc. 3, ser. 3. Washington DC: Armed Forces Institute of Pathology; 1992, p. 219-85.
Szulman AE, Surti U The syndromes of hydatiform mole II. Morphologic evolution of the complete and partial mole. Am J Obstet Gynecol. 1978; 132: 20-7.
Vassilakos P, Kajii T. Letter: Hydatidiform mole: two entities [letter]. Lancet 1976; 1: 259.
Vassilakos P, Riotton G, Kajii T. Hydatidiform mole: two entities. A morphologic and cytogenetic study with some clinical consideration. Am J Obstet Gynecol 1977; 127: 167-70.
OMS/WHO Scientific Group Gestational Trophoblastic Disease. World Health Organization Technical Reports Series 1983; p. 682.
Jacobs PA, Angell RR, Buchanan IM, Hassold TJ, Matsuyama AM, Manuel B. The origin of human triploids. Ann Hum Genet. 1978; 42: 49-57.
Jacobs PA, Wilson CM, Sprenkle JA, Rosenshein NB, Migeon BR. Mechanism of origin of complete hydatidiform moles. Nature 1980; 286: 714-6
Sheppard DM, Fisher RA, Lawler SD Karyotypic analysis and chromosome polymorphisms in four choriocarcinoma cell lines. Cancer Genet Cytogenet. 1985; 16: 251-8.
Scully RE, Young RH. Trophoblastic pseudotumor: a reappraisal. Am J Surg Pathol. 1981; 5: 75-6.
Finkler NJ, Berkowitz RS, Driscoll SG, Goldstein DP, Bernstein MR. Clinical experience with placental site trophoblastic tumors at the New England Trophoblastic Disease Center. Obstet Gynecol. 1988; 71(6 Pt 1): 854-7.
Twiggs LB, Okagaki T, Phillips GL, Stroemer JR, Adcock LL.Trophoblastic pseudotumor-evidence of malignant disease potential. Gynecol Oncol. 1981; 12(2 Pt 1): 238-48.
Abarca L, Madueño FJ, Alonso S et al. Tumor trofoblástico de asiento placentario. Descripción de 2 casos. Prog Obst Gin 1995; 28: 344-7.
Brewer CA, Adelson MD, Elder RC. Erythrocytosis associated with a placental-site trophoblastic tumor. Obstet Gynecol 1992; 79(5 Pt 2): 846-9.
Randall TC, Coukos G, Wheeler JE, Rubin SC. Prolonged remission of recurrent, metastatic placental site trophoblastic tumor after chemotherapy. Gynecol Oncol 2000; 76: 115-7.
Gillespie AM, Liyim D, Goepel JR, Coleman RE, Hancock BW. Placental site trophoblastic tumour: a rare but potentially curable cancer. Br J Cancer 2000; 82: 1186-90.
Prat J, Matias-Guiu X. Anatomía Patológica del aparato genital femenino. En: Mindán FJ, editor. Anatomía Patológica. Mosby; 1997; p. 939-77.
Miller DS, Ballon SC, Teng NNH. In: Endocrine disorders in pregnancy. Brody SA, Veland K, editors. Norwalk CT. Appleton & Lange 1989; p. 451.
Fisher RA, Newlands ES. Rapid diagnosis and classification of hydatidiform moles with polymerase chain reaction. Am J Obstet Gynecol. 1993; 168: 563-9.
Liber de arte medendi, De utero hydrope. Lib III, cap. XIII. Edición de Lyon 1564, p. 659.
Hertig AT, Mansell H. Tumors of the female sex organs. Part 1. Hydatiform mole and choriocarcinoma. En: Atlas of Tumor Pathology. (1st Series) Fascicle 33. Washington DC: Armed Forces Institute of Pathology, 1956.
Kajii T, Ohama K. Androgenetic origin of hydatidiform mole. Nature 1977; 268: 633-4.
Young RH, Kurman RJ, Scully RE. Placental site nodules and plaques. A clinicopathologic analysis of 20 cases. Am J Surg Pathol. 1990; 14: 1001-9.
Philippe E. Les maladies trofoblastiques gestationelles. Ann Pathol 1994; 14: 11-4
American College of Obstetrician and Gynecologist: Management of gestacional trophoblastic disease. Technical Bulletin No 178, March 1993 b.
Miller DS Lurain JR. Classification and staging of gestational trophoblastic tumors. Obstet Gynecol Clin North Am 1998; 15: 477.
Berkowitz RS, Hill JA, Kurtz CB, Anderson DJ. Effects of products of activated leukocytes (lymphokines and monokines) on the growth of malignant trophoblast cells in vitro. Am J Obstet Gynecol 1988; 158: 199-203.
Stellar MA, Mok SC, Yeh J, Fulop V, Anderson DJ, Berkowitz RS. Effects of cytokines on epidermal growth factor receptor expression by malignant trophoblast cells in vitro. J Reprod Med 1994; 39: 209-16.
Tomoda Y, Fuma M, Saiki N, Ishizuka N, Akaza T. Immunologic studies in patients with trophoblastic neoplasia. Am J Obstet Gynecol 1976; 126: 661-7.
Mogensen B, Kissmeyer-Nielsen F, Hauge M. Histocompatibility antigens on the HL-A locus in gestational choriocarcinoma. Transplant Proc 1969; 1: 76-9.
Berkowitz RS, Mostoufizadeh GhM, Kabawat SE, Goldstein DP, Driscoll SG. Immunopathologic study of the implantation site in molar pregnancy. Am J Obstet Gynecol 1982; 144: 925-30.
Lahey SJ, Steele G Jr, Berkowitz R, Rodrick ML, Ross DS, Goldstein DP, Zamcheck N, Wilson RE, Deasy JM. Identification of material with paternal HLA antigen immunoreactivity from purported circulating immune complexes in patients with gestational trophoblastic neoplasia. J Natl Cancer Inst 1984; 72: 983-90.
Mark FL, Ahearn J, Lathrop JC. Constitutional trisomy 8 mosaicism and gestational trophoblastic disease. Cancer Genet Cytogenet 1995; 80: 150-4.
Wolf NG, Lage JM. Genetic analysis of gestational trophoblastic disease: a review. Semin Oncol 1995; 22: 113-20.
Fisher RA, Newlands ES. Gestational trophoblastic disease. Molecular and genetic studies. Reprod Med 1998; 43: 87-97.
Kovacs BW, Shahbahrami B, Tast DE, Curtin JP. Molecular genetic analysis of complete hydatidiform moles. Cancer Genet Cytogenet 1991; 54: 143-52.
Roberts DJ, Mutter GL. Advances in the molecular biology of gestational trophoblastic disease. J Reprod Med 1994; 39: 201-8.
Sarkar S, Kacinski BM, Kohorn EI, Merino MJ, Carter D, Blakemore KJ. Demonstration of myc and ras oncogene expression by hybridization in situ in hydatidiform mole and in the BeWo choriocarcinoma cell line. Am J Obstet Gynecol 1986; 154: 390-3.
Cameron B, Gown AM, Tamimi HK.Expression of c-erb B-2 oncogene product in persistent gestational trophoblastic disease. Am J Obstet Gynecol 1994; 170: 1616-21.
Cheung AN, Srivastava G, Chung LP, Ngan HY, Man TK, Liu YT, Chen WZ, Collins RJ, Wong LC, Ma HK. Expression of the p53 gene in trophoblastic cells in hydatidiform moles and normal human placentas. J Reprod Med 1994; 39: 223-7.
Cheung AN, Srivastava G, Pittaluga S, Man TK, Ngan H, Collins RJ. Expression of c-myc and c-fms oncogenes in trophoblastic cells in hydatidiform mole and normal human placenta. J Clin Pathol 1993; 46: 204-7.
Fulop V, Mok SC, Genest DR, Gati I, Doszpod J, Berkowitz RS. p53, p21, Rb and mdm2 oncoproteins. Expression in normal placenta, partial and complete mole, and choriocarcinoma. J Reprod Med 1998; 43: 119-27.
Fulop V, Mok SC, Genest DR, Szigetvari I, Cseh I, Berkowitz RS. c-myc, c-erbB-2, c-fms and bcl-2 oncoproteins. Expression in normal placenta, partial and complete mole, and choriocarcinoma. J Reprod Med 1998; 43: 101-10.
Hashimoto K, Azuma C, Koyama M, Ohashi K, Kamiura S, Saji F. Loss of imprinting in choriocarcinoma [letter]. Nat Genet 1995; 9: 109-10.
Mutter GL, Stewart CL, Chaponot ML, Pomponio RJ. Oppositely imprinted genes H19 and insulin-like growth factor 2 are coexpressed in human androgenetic trophoblast. Am J Hum Genet 1993; 53: 1096-102.
Pattillo RA, Sasaki S, Katayama KP, Roesler M, Mattingly RF Genesis of 46, XY hydatidiform mole. Am J Obstet Gynecol. 1981; 141: 104-5.
Azuma C, Saji F, Tokugawa Y, Kimura T, Nobunaga T, Takemura M. Application of gene amplification by polymerase chain reaction to genetic analysis of molar mitochondrial DNA: the detection of anuclear empty ovum as the cause of complete mole. Gynecol Oncol. 1991; 40: 29-33.
Lawler SD, Povey S, Fisher RA, Pickthall VJ Genetic studies on hydatidiform moles. II. The origin of complete moles. Ann Hum Genet. 1982; 46(Pt 3): 209-22.
Ohama K, Kajii T, Okamoto E, Fukuda Y, Imaizumi K, Hagiwara K. Dispermic origin of XY hydatidiform moles. Nature. 1981; 292: 551-2.
Fishman DA, Padilla LA, Keh P, Cohen L, Frederiksen M, Lurain JR Management of twin pregnancies consisting of a complete hydatidiform mole and normal fetus. Obstet Gynecol. 1998; 91: 546-50.
Bristow RE, Shumway JB, Khouzami AN, Witter FR. Complete hydatidiform mole and surviving coexistent twin [review]. Obstet Gynecol Surv. 1996; 51: 705-9.
Puras A, Ezpeleta JM. Hallazgos morfológicos en la unidad feto-placentaria relacionados con la interrupción espontánea del embarazo. Rev Esp Patol 1998; 31: 237-246.
Lage JM, Mark SD, Roberts DJ, Goldstein DP, Bernstein MR, Berkowitz RS. A flow cytometric study of 137 fresh hydropic placentas: correlation between types of hydatidiform moles and nuclear DNA ploidy. Obstet Gynecol. 1992; 79: 403-10.
McFadden DE, Kwong LC, Yam IY, Langlois S Parental origin of triploidy in human fetuses: evidence for genomic imprinting. Hum Genet. 1993; 92: 465-9.
Rosai J. Akerman´s Surgical Pathology. 8th ed. St. Louis: Mosby, 1996.
Mutter GL, Pomponio RJ, Berkowitz RS, Genest DR Sex chromosome composition of complete hydatidiform moles: relationship to metastasis. Am J Obstet Gynecol. 1993; 168: 1547-51.
![]()