
Vol. 38, n.º 2, 2005
|
REVISTA
ESPAÑOLA DE
Vol. 38, n.º 2, 2005 |
CASUÍSTICA
Mauricio Corrales Santa1, Jorge Enrique Pinilla Pico2, Miguel Ignacio Roldán Pérez2, Jorge Iván Gutiérrez Montoya3
Universidad de Antioquia. Hospital Universitario San Vicente de
Paúl. Medellín. Colombia.
1 Cirugía Vascular.
2 Patología.
3 Cirugía de Trasplantes.
mauriciocorrales68@hotmail.com
RESUMEN
Antecedentes: El tumor neuroectodérmico primitivo (TNEP) es un tumor de células redondas que se presenta principalmente en tejidos blandos; excepcionalmente se encuentra en tracto intestinal, pelvis, retroperitoneo y riñón. Métodos: El espécimen quirúrgico renal, fue fijado en formol al 10%, e incluido en parafina. El estudio de inmunohistoquímica comprendió los siguientes anticuerpos: enolasa neuronal específica, sinaptofisina y mic2. Resultados: Reportamos el caso de un hombre de 32 años de edad atendido en el hospital universitario san Vicente de Paúl, Medellín con diagnóstico de tumor renal izquierdo, sin hematuria macroscópica al momento de ingreso ni metástasis a distancia documentadas, dolor de difícil manejo y sensación de masa abdominal a nivel del flanco izquierdo. Con dicho diagnóstico se lleva el paciente a cirugía para realizar nefrectomía radical, observando en el acto quirúrgico, gran compromiso ganglionar aortocavo y peripancreático. Sin ninguna posibilidad de ofrecer coadyuvancia de tipo radio o quimioterapia durante el post operatorio debido a su evolución clínica, fallece a los 2 meses postoperatorios con enfermedad diseminada. En el estudio anatomopatológico se evidencia proliferación renal difusa de células redondas con formación de rosetas, con infiltración de la cápsula hacia región abdominal, y ganglios locales comprometidos. El estudio de inmunohistoquímica resulta positivo para enolasa neuronal específica, sinaptofisina y mic2. Conclusiones: En los pacientes con diagnóstico de tumor renal, se debe llevar a cabo un estudio histopatológico completo, incluyendo el estudio de inmunohistoquímica antes de realizar una cirugía radical, debido al mal pronóstico de los TNEP, que pese a tener una baja frecuencia que conlleva a una mortalidad por encima del 80% (5). En la actualidad por medio del estudio inmunohistoquímico con sobre expresión del gene MIC2, detección de translocación especifica, estudios de hibridización con fluoresceína y trascripción reversa de polimerasa podemos diferenciar con certeza los TNEP primarios de los sarcomas de Ewing y otros sarcomas de células redondeadas.
Palabras Clave: tumor neuroectodérmico primitivo, tumor de Askin, sarcoma de Ewing, enolasa neuronal específica.
SUMMARY
Background: Primitive Neuroectodermal Tumor (PNET) is composed of small round cells and mainly found in soft tissues outside the central nervous system. However, urinary system is rarely involved. Patients and methods: A case of a 32 year old male complaining of severe pain in the left flank was diagnosed of a left renal tumor at the Hospital Universitario San Vicente de Paul, Medellin, Colombia. A palpable lump at left abdominal flank was noticed. There was no history of hematuria or documented metastatic lesions. Radical nephrectomy was performed. A diffuse enlargement of the left kidney with prominent lymph nodes around aortic, vena cava and pancreas were found. Because of the bad patient’s clinical condition, neither radio nor chemotherapy was offered. Patient died 2 months after surgery with disseminated metastases. Nephrectomy specimen was fixed in 10% formaldehyde and samples were paraffin embedded. An immunohistochemical study was performed including the following antibodies: NSE, synaptophysin and mic2. Results: Histological examination showed a diffuse renal proliferation of round cells with rosette formations. Invasion of the renal capsule and metastasis to lymph nodes were noticed. Immunohistochemical study reveals positivity for NSE, synaptophysin and mic2. Conclusions: In patients diagnosed of renal tumor, a complete hispathological study should be performed, including an immunohistochemical study before performing radical surgery. Although Primitive PNET is very infrequent, its mortality rate reaches 80%. Immunohistochemistry (mic2), specific translocation, flourescein and reversal transcription polimerase, can differentiate primary PNET from other round cell tumors.
Key words: neuroectodermic primitive tumor, PNET, Askin’s tumor, Ewing’s Sarcoma, neuron specific enolase.
NTRODUCCIÓN
En 1918 Stout definió a un grupo de neoplasias de células redondas de origen neuroectodérmico con localización variable como tumor neuroectodérmico primitivo (TNEP) (1), actualmente caracterizadas en la mayoría de los casos por una translocación cromosómica específica y reproducible t(11; 22) (q24; q12) (2,5,7), con diversos grados de diferenciación neuroectodérmica morfológica, inmunohistoquímica y ultraestructural; siendo este el segundo tipo de sarcoma más frecuente en las dos primeras décadas de vida (7).
El diagnóstico molecular se lleva a cabo demostrando los productos de los genes aberrantes, mediante la reacción en cadena de la polimerasa (7).
También se han descrito rearreglos del gen EWS en el cromosoma 22, demostrados por técnica de Fish, con tejidos incluidos en parafina (3).
El TNEP es una neoplasia maligna extremadamente agresiva, que se presenta con mayor frecuencia a nivel de tronco y región axial, confundiéndose con una masa derivada de hueso o tejidos blandos. Aquellos de origen renal y en general retroperitoneal son raros y en la literatura se encuentran publicados como reportes aislados, tanto en adolescentes como en adultos jóvenes, siendo estas las edades de mayor incidencia.
La agresividad y el desenlace fatal de ésta patología requiere un diagnóstico temprano y acertado, diferenciándolo de algunas entidades clínicas similares como: neuroblastoma (segunda neoplasia sólida extracraneal más común), sarcoma de Ewing’s, tumor de Wilms, neuroepitelioma y otros tumores de células redondas.
Para la publicación de éste caso se revisó tanto la presentación clínica como los hallazgos radiológicos e histopatológicos, mostrando finalmente el resultado.
CARACTERÍSTICAS PATOLÓGICAS
Los TNEP resecados son grandes, pálidos, blandos y con extensa necrosis (4).
En los tumores axiales, suele ser difícil definir si se origina de hueso o de tejidos blandos (4).
Microscópicamente, presentan un espectro de diferenciación neuro-ectodérmica, siendo constante un patrón lobular o trabecular, muy vascularizado, con escaso estroma, y focos de necrosis (fig. 1). En el extremo pobremente diferenciado, sarcoma de Ewing, las células tumorales presentan citoplasma escaso y pálido, núcleo redondo u ovoide con cromatina muy fina; la presencia de nucleolo es variable. En el extremo mejor diferenciado, neuroepitelioma, las células presentan un citoplasma eosinófilo, cromatina gruesa, nucléolo, numerosas rosetas de tipo Homer-Wright y pseudo rosetas peri vasculares(4) (fig. 2).
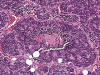
Fig. 1: Se
Observan focos con patrón lobular, en medio del cual se encuentran extensas
zonas de necrosis.
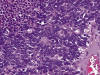
Fig. 2: En
este ejemplo se observan escasas rosetas tipo Homer Wright.
El extremo indiferenciado es con mayor frecuencia positivo al ácido peryódico de Schiff (PAS) (4).
Desde el punto de vista inmunohistoquímico, el tumor es positivo para el antígeno CD99, el cual debe ser interpretado dentro del contexto de los tumores de células redondas debido a que los linfomas T linfoblásticos, sarcomas sinoviales pobremente diferenciados, carcinomas neuroendocrinos y los rabdomiosarcomas de tipo alveolar también pueden mostrar positividad. Muchos TNEP son positivos para los antígenos: enolasa neuronal específica, PGP 9.5, neurofilamentos, LEU 7, sinaptofisina y MIC2 (4-6). El 10% de los TNEP son positivos para citoqueratina (4). Otros marcadores de inmunohistoquímica con expresión variable son: vimentina, S-100, cromogranina, Trk (8).
Hay un grupo de TNEP que presenta diferenciación poli fenotípica, con diferenciación glial, ganglionar o epitelial, como resultado de la pluripotencialidad de la cresta neural (4).
La familia de los tumores neuroectodérmicos primitivos periféricos incluyen varios tumores que muestran características propias del neuroepitelio germinal o de la cresta neural en desarrollo (tabla 1) (6).

REPORTE DEL CASO
Un hombre de 32 años de edad atendido en el hospital universitario San Vicente de Paúl, Medellín con dolor en el flanco izquierdo, evidente masa local de 15 ´ 20 cm por palpación, firme, fija y con extensión a la pelvis. Perdida de 5 kg de peso en los 2 últimos meses sin cambios en su hábito intestinal y sin clínica de compromiso vascular.
Los resultados de ayudas diagnósticas incluyen: Rayos X de tórax interpretados como normales, excepto por una leve elevación del hemidiafragma izquierdo. La tomografía helicoidal reveló una gran masa renal izquierda, heterogénea que reemplazaba casi totalmente el riñón ipsilateral, con invasión ganglionar peri aórtica y pancreática distal con desplazamiento derecho de estos órganos sin mostrar evidencia de compromiso hepático o resto de estructuras intra abdominales.
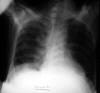
Fig. 3: Rayos
X de Tórax Paciente con TNEP prequirúrgico.
El paciente es llevado a cirugía con diagnóstico de tumor renal primario compatible con hipernefroma como primera impresión, dado el compromiso encontrado en la tomografía y el dolor ocasionado, motivo principal de consulta.
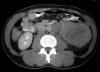
Fig. 4: TAC
abdominal masa renal Izquierda.
Se realiza un abordaje lumbar izquierdo postero lateral, encontrando una gran masa de 16 ´ 12 ´ 14 cm que sobrepasa la cápsula renal e invade tejidos adyacentes; psoas izquierdo, diafragma y ganglios periféricos aortocavo y peripancreáticos descritos. Respetando bazo, glándula suprarrenal y tubo digestivo. Se realiza una nefrectomía radical que incluyó vaciamiento de ganglios comprometidos y resección parcial del hemidiafragma izquierdo, marcando lecho quirúrgico con endoclip para posible terapia coadyuvante posterior.
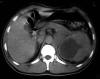
Fig. 5: TAC
abdominal compromiso total riñón izquierdo.
Evolución posterior normal y alta al 5.º día post quirúrgico en buenas condiciones. Una cita ambulatoria de control reporta proceso cicatrizal adecuado, sin embargo reingresa un mes después por presentar dificultad respiratoria, agitación y desorientación. Control tomográfico demuestra derrame pleural bilateral con lesiones nodulares pulmonares y peri aórticas bilaterales compatibles con metástasis, además imágenes nodulares retroperitoneales y ascitis importante.
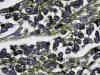
Fig. 6: CD
99 (MIC 2): En esta coloración de inmunohistoquímica se observa positividad de
membrana citoplasmática.
Su evolución posterior no permitió brindar ninguna terapia coadyuvante tipo radiación o quimioterapia y por tanto el paciente solamente recibe manejo paliativo sintomático en casa durante dos meses antes de su muerte.
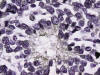
Fig. 7: Sinaptofisina:
Con esta coloración se observa positividad con tinción citoplasmática. Enolasa
neuronal específica: Con esta coloración se observa positividad con tinción
citoplasmática.
MÉTODOS
El tejido fue fijado en formol al 10%, y procesado para histología del modo usual. La inmunohistoquímica fue realizada en tejidos incluidos en parafina. El panel de anticuerpos comprendió: enolasa neuronal específica-M0873 (DAKO, Carpintería, California, EE.UU.); sinaptofisina-A0010 (DAKO, Carpintería, California, EE.UU.); y mic2-M3601 (DAKO, Carpintería, California, EE.UU.).
RESULTADOS
Patología Macroscópica
El espécimen consistió del riñón izquierdo, con peso de 1781 g, con medidas de 20 ´ 15 ´ 14 cm, rodeado de tejido de aspecto graso, con fragmento de uréter de 2 ´ 0,7 cm, y hacia el polo superior con fragmentos de glándula suprarrenal. La superficie normal del riñón fue distorsionado por múltiples nódulos, congestivos.
Al corte, solo se observan 2 cm de parénquima renal conservado, el tejido restante correspondía a un extenso nódulo tumoral color blanco y gris, con múltiples zonas de aspecto hemorrágico y necrótico reemplazando virtualmente el riñón, y ocupando el sistema calicial.
Histopatología
La muestra corresponde a tejido tumoral formado por abundantes células con marcada alteración de la relación núcleo/citoplasma, con núcleos homogéneos, hipercromáticos, pérdida de la polaridad, cromatina grumosa, frecuentes mitosis aberrantes, nucleolos prominentes, e infiltración de las paredes circundantes, a modo de cordones, formando pseudorrosetas perivasculares y rosetas tipo Homer Wright, con extensas zonas de necrosis. El parénquima renal conservado, presenta gran infiltración por células neoplásicas descritas, las cuales también se ubican en zona perirrenal, con dilatación tubular y presencia de infiltrado inflamatorio mononuclear.
Presenta infiltración neoplásica perivascular a nivel de hilio renal; infiltración difusa de la glándula suprarrenal, y reemplazo total de la arquitectura de los ganglios linfáticos.
Las suprarrenales no muestran evidencias de neuroblastoma o cambios regresivos que sugirieran la temprana presencia de un neuroblastoma.
Las tinciones inmunohistoquímicas para enolasa neuronal específica, sinaptofisina y MIC 2 son positivas.
DISCUSIÓN
En este artículo, nosotros reportamos el caso de un paciente de sexo masculino, de 32 años de edad, con diagnóstico clínico y radiológico de tumor renal, nefrectomizado, al examen histopatológico clasificado dentro del grupo de los tumores de células pequeñas, redondas y azules; con tinciones de inmunohistoquímica positivas para enolasa neuronal específica, sinaptofisina y MIC 2, por lo cual se diagnostica como tumor neuroectodérmico primitivo renal. Falleciendo a los 2 meses de realizado el diagnóstico y procedimiento.
Las series actuales sugieren que el diagnóstico de TNEP renal debe ser considerado en niños y adolescentes o adultos jóvenes con neoplasias renales avanzadas, con metástasis locales o a distancia, y una pobre respuesta al tratamiento (5,7). Se revisan 5 casos reportados en la literatura, tratados con terapia combinada, quimioterapia y radioterapia (2), y solo quimioterapia (3), encontrando una mortalidad del 60%, de ellos 2 de terapia combinada, y 1 de terapia simple (5).
En cuanto al diagnóstico, es importante diferenciar el TNEP renal de otras patologías como: tumor de Wilms, angiomiolipoma, linfoma maligno, entre otros, por cuanto el abordaje terapéutico y los resultados del mismo son bien diferentes en estas entidades.
Desde el punto de vista histopatológico, el TNEP renal debe ser considerado ante el diagnóstico de tumor de Wilms en un adulto, ya que estos dos tumores se presentan con mayor frecuencia en adultos jóvenes (10,11). Además el tumor de Wilms de tipo blastemal también puede ser positivo para CD 99 (9).
El TNEP puede ser histológicamente indistinguible del neuroblastoma renal clásico, el cual también presenta un comportamiento agresivo, con enfermedad extensa al momento del diagnóstico (12-14).
En algunos casos, en los que es difícil diferenciarlo del Tumor rabdoide maligno (TRM),-17 son las anormalidades citogenéticas el factor determinante (5). Weeks y cols. reportaron en una revisión histopatológica, de 56 neoplasias renales que semejaban ser TRM, un total de 8 casos de TNEP renal (5).
Parham y cols. encontraron en una revisión de 42 casos diagnosticados como TRM extrarrenales, 4 tumores de origen neuroectodérmico (5).
El curso clínico de TNEP renal es similar al del TRM, con pobre respuesta al tratamiento y una mortalidad del 80%. Sin embargo, el TRM suele ocurrir durante los 2 primeros años de vida (5).
Es probable que los TRM y TNEP se originen en las mismas células primitivas, lo que explicaría su comportamiento clínico similar (5).
Basados tanto en el caso acá reportado, como en los casos reportados en la literatura, nosotros consideramos que el TNEP renal constituye una entidad clínica única, diferente del TNEP de otras localizaciones, como tracto intestinal, pelvis, o retroperitoneo. Con un comportamiento clínico más agresivo que estos últimos, y en algunos casos similar al del TRM, y tumor de Wilms. Siendo desde el punto de vista histopatológico esencialmente un tumor de células pequeñas, redondas y azules, por lo cual se deben realizar estudios citogenéticos, con detección de la translocación especifica, estudios de hibridización con fluoresceína, trascripción reversa de polimerasa, y estudio inmunohistoquímico para CD99, en cualquier paciente en el cual sea considerado este diagnóstico. (15).
BIBLIOGRAFÍA
Stout AP. A tumor of the ulnar nerve. Proc NY Pathol Soc 1918; 18: 2-12.
Thiele CJ. Pediatric peripheral neuroectodermal tumors, oncogenes, and differentiation. Cancer Invest 1990; 8: 629-39.
Delattre O, Zucman J, Melot T, Sastre Garau X, Zucker JM, Lenoir GM, et al. The Ewing family of tumors. A subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331: 294-9.
Fletcher C, editor. Diagnostic histopathology of tumors. 2.ª ed. 2000; 1698-700.
Rodríguez-Galindo C, Marina N, Fletcher B, Parham D, Bodner S, Meyer W. Is Primitive Neuroectodermal Tumor of the Kidney a Distinct Entity? Cancer 1997; 79: 11 2243-9.
D’Amore E, Ninfo V. Soft Tissue Small Round Cell Tumors: Morphological Parameters. Sem Diag Pathol 1996; 13: 184-203.
Marley E, Liapis H, Humphrey P, Nadler R, Siegel C, Zhu X, Brandt J, Dehner L. Primitive Neuroectodermal Tumor of the Kidney- Another Enigma: a Pathologic, Immunohistochemical, and Molecular Diagnostic Study. Am J Surg Pathol 1997; 21: 354-9.
Llombart-Bosch A, Contesso G, Peydro-Olaya A. Histology, Immunohistochemistry, and Electron Microscopy of Small Round Cell Tumors of Bone. Semin Diag Pathol 1996; 13: 153-70.
Dabbs DJ, editor. Diagnostic Inmunohistochemistry. Editorial Churchill Livingstone: 2002. p. 73.
Kattan J, Tournade. MF, Culine S, Terrier-Lacombe Mj, Droz Jp. Adult Wilms tumour. Review of 22 cases. Eur J Cancer 1994; 30A: 1778-82.
Arrigo S, Beckwith J, Sharples K, Dángio G, Haase G. Better survival after combined modality care for adults with Wilms tumor. A report from the National Wilms tumor Study. Cancer 1990; 66: 827-30.
Rosenfield NS, Leonidas JC, Barwick KW. Aggresive neuroblastoma simulating Wilms tumor. Radiology 1988; 166: 165-7.
Gohji K, Nakanishi T Hara I, Hamami G, Kamidona S. Two cases of primary neuroblastoma of the kidney in adults. J Urol 1987; 137: 966-8.
Sinoda M, Tsutsumi Y, Hata J, Yokohama S. Peripheral neuroepithelioma of childhood. Immunohistochemical demonstration of epithelial differentiation. Arch Pathol Lab Med 1998; 112: 1155-8.
Pappo AS, Douglass EC, Meyer WH, Marina NM, Paham DM. Use of HBA-71 and anti beta-2-microblobulin to distinguish peripheral neuroepithelioma from neuroblastoma. Hum Pathol 1993; 24: 880-5.
![]()